Introduzione
L’anemia mediterranea, o beta-talassemia, è una malattia ereditaria del sangue caratterizzata da una ridotta quantità di emoglobina e globuli rossi circolanti nel sangue (l’emoglobina è una sostanza che si trova nei globuli rossi che consente a queste cellule di trasportare ossigeno).
Nei soggetti colpiti da beta talassemia i sintomi possono essere da quasi nulli a molto gravi, a seconda della forma di cui si è portatore: da un punto di vista generale i bassi livelli di emoglobina (anemia) causano una carenza di ossigeno generalizzata, dovuta alla riduzione del numero di globuli rossi circolanti, che si traduce in diversi possibili sintomi.
- pallore,
- debolezza,
- fatica
e talvolta complicazioni più severe, come
- un’aumentata suscettibilità alle infezioni,
- un aumento della dimensione della milza,
- un sovraccarico di ferro,
- …
L’anemia mediterranea è classificata in base alla gravità della forma, che può colpire in tre modi:
- major,
- intermedia,
- minor.
I sintomi nei bambini affetti da talassemia major in genere compaiono entro i due anni di vita in tutta la loro gravità, richiedendo un rapido intervento medico attraverso trasfusioni di sangue (che presumibilmente dovranno periodicamente essere ripetute per tutta la vita).
La forma minor è invece decisamente più lieve e nella maggior parte dei casi non richiede alcun trattamento, se non un regolare monitoraggio dei livelli di ferro per evitare pericolosi accumuli.
La forma intermedia è appunto una manifestazione che si pone a metà strada, richiedendo occasionali trasfusioni di sangue.
La diagnosi avviene attraverso l’esecuzione di specifici esami del sangue, mentre il trattamento (necessario solo nelle forme più severe) prevede tra l’altro trasfusioni di sangue e rimozione del ferro in eccesso dall’organismo. Nei soggetti portatori con sintomi lievi o nulli non è in genere necessaria alcuna terapia, anche se un corretto stile di vita è fondamentale per mantenere uno stato di benessere, ad esempio seguendo una buona dieta che contribuisca a rafforzare la propria energia.
Portatore sano
Relativamente all’anemia mediterranea ciascun individuo può essere classificato in uno dei tre modi seguenti:
- sano,
- malato,
- portatore sano.
Il soggetto sano non mostra alcun sintomo, non ha legami con la condizione e non può in alcun modo trasmetterne i geni ai propri figli.
Il soggetto malato può trasmettere ai figli sono geni portatori della malattia (geni malati)
Il portatore sano di talassemia è il paziente che mostra nel codice genetico solo uno dei geni difettosi che causano la malattia (uno su due) e non ne mostra i sintomi caratteristici (se non in minima parte); per questa ragione in questi individui si parla spesso di condizione, più che di malattia, in quanto in grado di trasmettere ai figli sia un gene malato che uno sano.
La condizione della prole è quindi determinata da entrambi i genitori.

iStock.com/Gam1983
Cause
La causa della talassemia è la presenza di difetti nel codice genetico coinvolto nella sintesi di emoglobina: l’unico modo per sviluppare la talassemia è ereditare uno o più geni di emoglobina difettosi dai propri genitori.
L’emoglobina è una proteina rossa, ricca di ferro, presente nei globuli rossi che consente a quest’ultimi di trasportare l’ossigeno dai polmoni a tutte le parti del corpo e, viceversa, l’anidride carbonica proveniente dalle altre zone dell’organismo ai polmoni in modo che possa essere eliminato.
La maggior parte delle cellule del sangue, compresi i globuli rossi, sono prodotti regolarmente nel midollo osseo, un materiale rosso e spugnoso che si trova all’interno della cavità di molte delle ossa di grandi dimensioni. L’anemia mediterranea riduce la produzione di emoglobina ed aumenta il tasso di distruzione dei globuli rossi nel sangue, causando così anemia. Quando si è anemici il sangue non contiene abbastanza globuli rossi da riuscire a trasportare la giusta quantità di ossigeno a tutto il corpo, determinando fra l’altro una sensazione di affaticamento.
Ci sono due tipi principali di talassemia, alfa e beta, dal nome delle due catene proteiche dell’emoglobina che possono essere affette dall’errore genetico:
- in Africa è più diffusa l’alfa talassemia,
- mentre nel bacino del Mediterraneo è più diffusa la beta talassemia (da un punto di vista di definizioni la vera anemia mediterranea).
Il tipo di anemia di cui si soffre dipende dal tipo di difetto genetico che si eredita dai genitori.
Alfa-talassemia
Quattro geni sono coinvolti nella sintesi della catena alfa dell’emoglobina, e di questi se ne ottengono due da ciascun genitore; se uno o più geni alfa dell’emoglobina sono difettosi allora si sviluppa l’alfa-talassemia.
La gravità dei sintomi dipende dal numero di alleli (geni) coinvolti, passando da
- una condizione del tutto asintomatica (delezione di un allele, si parla di portatore silente),
- a una incompatibile con la vita (delezione di tutti e quattro e morte del feto durante la gravidanza).
Beta-talassemia
Si tratta della forma più comune in Italia, soprattutto nel meridione; due geni sono coinvolti nella sintesi della catena beta dell’emoglobina e se ne ottiene uno da ciascun genitore. Se uno o entrambi i geni sono difettosi si sviluppa la beta-talassemia.
Le possibili alterazioni a livello del codice genetico possono causare
- la riduzione (beta+)
- o l’assenza (beta0)
della sintesi delle catene beta dell’emoglobina, da cui risultano le seguenti possibili condizioni:
- Talassemia minor, si verifica quando solo uno degli alleli porta la mutazione della malattia (un gene è sano, l’altro è beta+ o beta0). I sintomi sono minimi.
- Talassemia intermedia, si verifica quando entrambi gli alleli sono alterati, ma non nel modo più grave possibile (entrambi i geni sono beta+, oppure un gene è beta+ e l’altro beta0). Il paziente potrebbe richiede occasionali trasfusioni.
- Talassemia major, si eredita quando entrambi gli alleli sono mutati (beta0) ed è causa delle manifestazioni più gravi; richiede tassativamente periodiche trasfusioni di sangue.
Fattori di rischio
La malattia è ereditaria, quindi il rischio nasce se uno o entrambi i genitori sono portatori della mutazione genetica:
- la beta talassemia è più comune nelle persone di origine italiana, greca, medio-orientale, sud-asiatica ed africana.
- l’alfa-talassemia colpisce soprattutto le persone del sud-est asiatico, di discendenza cinese e filippina.
Sintomi
I sintomi sono più o meno accentuati in base al tipo di talassemia ereditato, nel caso della forma minor in genere sono del tutto assenti.
Alla luce del fatto che nell’organismo del soggetto colpito da anemia mediterranea ci sono meno globuli rossi del normale i sintomi, quando presenti, sono quelli tipici dell’anemia (livelli di emoglobina insufficienti), tra cui:
- vertigini,
- mancanza di fiato,
- battito cardiaco accelerato,
- mal di testa,
- crampi alle gambe,
- difficoltà di concentrazione,
- pallore.
Un bimbo che erediti la forma intermedia o major entro i due anni di vita potrebbe manifestare:
- debolezza e affaticamento,
- pallore o ittero (pelle di colore giallo dovuto all’accumulo di bilirubina),
- irritabilità,
- deformità ossee del viso,
- rallentato sviluppo,
- addome sporgente,
- urine scure,
- malocclusione dentale.
Diagnosi
La diagnosi di anemia mediterranea si basa oggi su un protocollo clinico consolidato che combina l’analisi della storia familiare con esami di laboratorio di precisione. Il percorso diagnostico è fondamentale non solo per chi presenta sintomi, ma anche per identificare i portatori sani che potrebbero trasmettere la condizione alla prole.
Esami del sangue di primo livello
Il primo sospetto nasce solitamente da un normale emocromo. Nei soggetti con talassemia, si osserva tipicamente una microcitosi, ovvero globuli rossi di dimensioni ridotte (basso valore di MCV), associata a una riduzione dei livelli di emoglobina. A differenza dell’anemia da carenza di ferro, il numero totale di globuli rossi può risultare normale o addirittura superiore alla norma.
Per distinguere la talassemia da una carenza marziale, il medico prescrive il dosaggio di:
- Ferritina e transferrina: per valutare le riserve di ferro nell’organismo.
- Sideremia: per misurare il ferro circolante.
Elettroforesi dell’emoglobina e test molecolari
Il “gold standard” per la diagnosi è l’elettroforesi dell’emoglobina o, più comunemente oggi, la cromatografia liquida ad alta prestazione (HPLC). Questi test permettono di quantificare i diversi tipi di emoglobina presenti (come l’emoglobina A2 e l’emoglobina F). Nei portatori di beta-talassemia minor, si riscontra tipicamente un aumento della frazione HbA2.
L’analisi del DNA (test molecolari) rappresenta lo step definitivo. Viene utilizzata per identificare le specifiche mutazioni genetiche responsabili della malattia. Questo esame è cruciale per definire la prognosi nelle forme intermedie e per lo screening di coppia in fase preconcezionale.
Diagnosi prenatale e screening
Per le coppie a rischio (entrambi portatori sani), sono disponibili protocolli di diagnosi prenatale per identificare precocemente la condizione del feto:
- Villocentesi: eseguita solitamente tra la 10ª e la 12ª settimana di gestazione tramite il prelievo di villi coriali.
- Amniocentesi: effettuata dopo la 15ª settimana tramite il prelievo di liquido amniotico.
- Diagnosi genetica pre-impianto (PGT): opzione disponibile per le coppie che ricorrono alla fecondazione assistita, che permette di analizzare l’embrione prima del trasferimento in utero.
Pericoli
L’organismo del paziente affetto da una forma severa di talassemia proverà a compensare la situazione sforzandosi di produrre più globuli rossi. Uno dei siti di produzione dei globuli rossi è il midollo osseo, un tessuto specializzato all’interno delle ossa, che venendo così fortemente stimolato avrà come conseguenza la formazione di ossa allungate e quindi più fragili, più inclini alla rottura (osteoporosi).
L’altro sito di produzione è la milza, un organo che si trova nel lato sinistro dell’addome e che svolge anche numerosi altri compiti, tra cui aiutare il sistema immunitario a proteggere l’organismo dalle infezioni.
Il paziente talassemico andrà quindi incontro a un ingrandimento della milza sottoposta a eccessive richieste di sintesi di globuli rossi, ma distogliendo così l’attenzione dagli altri suoi compiti; verrà abbassata la guardia causando un pericoloso calo delle difese immunitarie, che esporrà l’organismo al rischio di infezioni.
Altro rischio che corre il paziente è quello di eccessivo accumulo di ferro a causa della dieta o soprattutto a seguito delle ripetute trasfusioni; i rischi sono:
- cardiaci,
- epatici,
- endocrino,
- pubertà ritardata,
- bassi livelli di estrogeni (ormoni femminili) e testosterone (ormone maschile),
- diabete,
- problemi con la tiroide (ipotiroidismo) e paratiroidi (ipoparatiroidismo).
La talassemia major può inoltre essere causa di:
- ritardi di crescita,
- calcoli biliari che possono causare l’infiammazione della cistifellea (colecistite),
- dolore addominale,
- ittero,
- crescite ossea anomale,
- osteoporosi,
- riduzione della fertilità.
Dieta
I pazienti non trasfusi affetti da talassemia intermedia sono incoraggiati a seguire un’alimentazione povera di ferro e a bere tè durante i pasti, che diminuisce l’assorbimento intestinale del ferro.
I bambini talassemici e trasfusi sono ancora relativamente anemici, per cui l’organismo potrebbe ancora richiedere il ferro, in quantità da valutare con l’aiuto del pediatra. Per quanto possa essere difficile seguire da vicino la loro dieta, essi dovrebbero sviluppare buone abitudini sin da subito. Ricordate inoltre che il ferro contenuto nella carne è viene più facilmente assorbito rispetto a quello proveniente da fonti vegetali.
Non cucinate con pentole in ghisa in quanto il ferro degli utensili da cucina si può trasmettere al cibo. Alcuni alimenti, come il succo d’arancia, possono migliorare l’assorbimento del ferro, mentre altri come tè, latte e caffè, possono ridurne l’assorbimento.
Possono essere utili le integrazioni con vitamina D, calcio e acido folico (rispettivamente per la prevenzione dell’osteoporosi i primi due, per la produzione di globuli rossi il terzo).
Salvo che il medico non lo prescriva, si raccomanda di evitare l’assunzione di vitamine o altri integratori contenenti ferro.
Cura e trattamento
L’approccio terapeutico all’anemia mediterranea ha vissuto una trasformazione radicale negli ultimi anni. Se un tempo l’obiettivo era esclusivamente la sopravvivenza, oggi la medicina punta a garantire una qualità della vita paragonabile a quella della popolazione generale e, in casi selezionati, alla guarigione definitiva. Gli obiettivi principali della terapia sono il mantenimento di livelli adeguati di emoglobina, la prevenzione dei danni d’organo da accumulo di ferro e la correzione del difetto genetico sottostante.
Gestione della talassemia trasfusione-dipendente
Per i pazienti con talassemia major o forme intermedie gravi, il pilastro del trattamento rimane la terapia trasfusionale regolare. Le trasfusioni vengono solitamente effettuate ogni 2-4 settimane per mantenere i livelli di emoglobina pre-trasfusionale tra 9,5 e 10,5 g/dL. Questo approccio permette di sopprimere l’eritropoiesi inefficace (la produzione difettosa di sangue) e prevenire le deformazioni ossee.
La conseguenza inevitabile delle trasfusioni croniche è il sovraccarico di ferro. Per contrastarlo, si utilizza la terapia chelante. I farmaci chelanti legano il ferro in eccesso e ne favoriscono l’eliminazione attraverso le urine o le feci. Oggi sono disponibili opzioni orali (come il deferasirox o il deferiprone) molto più gestibili rispetto alle vecchie infusioni sottocutanee notturne, migliorando significativamente l’aderenza del paziente al trattamento.
Nuove terapie farmacologiche
Una delle innovazioni più recenti è l’introduzione di farmaci che stimolano la maturazione dei globuli rossi (agenti di maturazione eritroide come il luspatercept). Questi medicinali possono ridurre significativamente il fabbisogno trasfusionale in molti pazienti adulti, alleviando il carico terapeutico e riducendo l’accumulo di ferro.
Opzioni curative: trapianto e terapia genica
Esistono oggi due strade per una guarigione definitiva:
- Trapianto di midollo osseo (o di cellule staminali emopoietiche): è l’opzione curativa standard, più efficace se eseguita in età pediatrica e con un donatore compatibile (solitamente un fratello). Sebbene molto efficace, comporta rischi significativi che vanno valutati attentamente con l’equipe medica.
- Terapia genica: rappresenta la frontiera più avanzata. Consiste nel prelevare le cellule staminali del paziente stesso, correggerne il difetto genetico in laboratorio (tramite vettori virali o tecniche di editing genomico come CRISPR/Cas9) e reinfonderle nel paziente. Questo approccio elimina il rischio di rigetto e permette potenzialmente ai pazienti di diventare indipendenti dalle trasfusioni per tutta la vita.
Stile di vita e prevenzione
Oltre alle terapie mediche, il paziente talassemico deve adottare strategie quotidiane per preservare la propria salute:
- Esercizio fisico: Una regolare attività fisica di intensità moderata è fondamentale per proteggere la salute delle ossa e migliorare la capacità cardiovascolare, riducendo il rischio di osteoporosi e stanchezza cronica.
- Prevenzione delle infezioni: A causa della vulnerabilità immunitaria (spesso legata alla splenectomia o al sovraccarico di ferro), è essenziale seguire scrupolosamente il calendario vaccinale, inclusi i vaccini influenzali, pneumococcici e per l’epatite B.
- Monitoraggio multidisciplinare: Controlli regolari con cardiologi, endocrinologi ed epatologi sono necessari per individuare precocemente eventuali complicanze legate al ferro.
Anemia mediterranea e figli
Essendo una malattia ereditaria non è possibile prevenire la trasmissione della malattia, se non ricorrendo a forme di riproduzione assistita che combinino l’impianto di pre-diagnosi genetica con la fecondazione in vitro.
Si tratta di un moderno approccio in grado di aiutare i genitori affetti da talassemia o che sono portatori di geni di emoglobina difettosi, a dare alla luce bambini sani.
La procedura prevede il recupero di uova mature da una donna e la fecondazione di questi tramite lo sperma di un uomo contenuto in una provetta di laboratorio; gli embrioni sono testati per i geni difettosi e soltanto quelli privi di difetti vengono impiantati nella donna.
Nel caso di portatori sani è quindi opportuno procedere alla verifica del partner, se questi risultasse non portatore i figli della coppia (portatore – non portatore) avrebbero ad ogni concepimento:
- 50% di probabilità di essere a loro volta portatori,
- 50% di probabilità di essere perfettamente sani.
Nel caso in cui un genitore fosse sano e l’altro malato i figli sarebbero sicuramente portatori del tratto talassemico.
Se entrambi fossero portatori il bambino avrebbe:
- 25% di probabilità di essere perfettamente sano;
- 50% di probabilità di essere a sua volta portatore;
- 25% di probabilità di nascere con una forma grave.
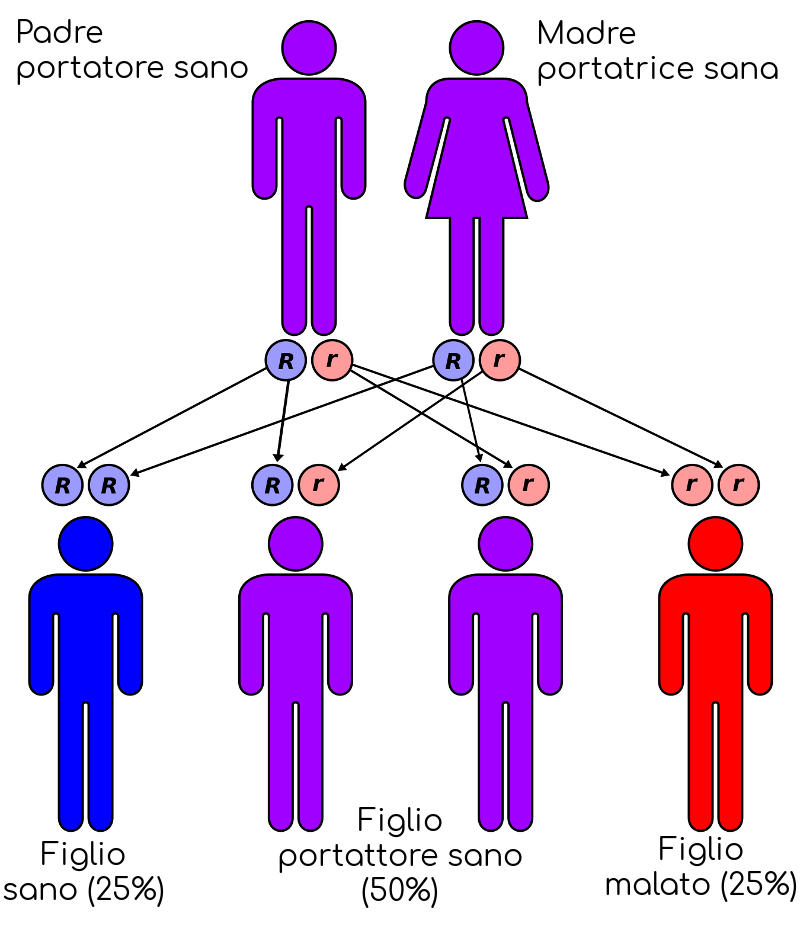
By en:User:Cburnett – Own work in Inkscape, CC BY-SA 3.0, Link
Si tratta in ogni caso di semplificazioni, in quanto esempi che non tengono conto del tipo di alterazione genetica dei genitori (beta0 o beta+) e per questo si raccomanda in caso di dubbi di fare il punto con un genetista.
Gravidanza e fertilità
Per fertilità s’intende la capacità di avere e portare a termine una gravidanza: una ridotta fertilità è comune agli individui affetti da anemia mediterranea dipendente da trasfusioni, come la beta-talassemia maggiore. Mentre alcuni pazienti potrebbero essere del tutto sterili, altri invece potrebbero riuscire ad avere figli biologici tramite fecondazione assistita o altri interventi medici.
Alcuni pazienti sono infine assolutamente in grado di concepire anche senza intervento medico.
Cause di riduzione della fertilità
La fertilità è influenzata dalla capacità degli ovuli femminili e dello sperma maschile di maturare ed unirsi nella fecondazione: nelle donne è influenzata anche dalla maturazione sessuale e dalla capacità dell’utero di portare a termine una gravidanza. Un ritardo nella maturazione sessuale può escludere la possibilità di avere figli biologici finchè la pubertà non sia raggiunta e, per le ragazze, fino alla comparsa del primo ciclo mestruale (menarca). Alcune donne con la beta talassemia soffrono di amenorrea primaria (cioè il ciclo mestruale non è mai iniziato), condizione che può richiedere la somministrazione di terapie ormonali. Lo stesso vale per l’amenorrea secondaria, in cui una donna comincia ad un certo punto a non avere più le mestruazioni.
Una riduzione della fertilità nei soggetti affetti da anemia mediterranea è principalmente attribuibile all’eccesso di ferro in uno o più organi o nelle ghiandole che contribuiscono alla produzione di ovuli o sperma:
- nelle donne gli ovuli nelle ovaie maturano dietro l’azione ormonale guidata dall’ipofisi, che riceve ordine di produrre specifici ormoni (o di stopparne la produzione) dall’ipotalamo, a sua volta legato a doppio filo alle ovaie;
- negli uomini lo sperma viene prodotto nei testicoli. Come per le ovaie, anche i testicoli ricevono i segnali ormonali della ghiandola pituitaria, che a sua volta riceve i segnali dall’ipotalamo.
La fertilità può essere ridotta a causa di un eccesso di ferro nella ghiandola pituitaria (ipofisi): il danno che ne deriva impedisce il rilascio di ormoni ipofisari in risposta ai segnali provenienti dall’ipotalamo. L’eccesso di ferro può verificarsi anche nell’ipotalamo.
L’infertilità può sopraggiungere anche quando un sovraccarico di ferro nelle ovaie o nei testicoli provoca danni alle cellule delle uova o degli spermatozoi.
Prevenire la riduzione della fertilità
Sembra che l’approccio migliore per cercare una gravidanza quando si ha l’anemia mediterranea consista nel tenere sotto controllo i livelli di ferro: il corpo non è in grado di sbarazzarsi delle quantità in eccesso che si accumulano per via delle trasfusioni croniche di sangue. La desferrioxamina (Desferal) aiuta a rimuovere l’eccesso di ferro. Questo farmaco viene in genere somministrato cinque notti su siete a settimana tramite una pompa che infonde lentamente la desferrioxamina sotto la pelle per diverse ore. Studi scientifici suggeriscono che un uso efficace della desferrioxamina può condurre ad una regolare maturazione sessuale. I pazienti che agiscono al meglio sono quelli che cominciano precocemente il trattamento, prima che i livelli di ferro, misurati in termini di incremento del livello di ferritina, diventino troppo elevati. Coloro che mantengono basso il livello di ferro nel corso del trattamento sembrano anche avere una maggiore possibilità di preservare la loro fertilità.
Tuttavia, anche coloro con livelli di ferritina molto elevati per un lungo periodo di tempo, potrebbero avere una maturazione sessuale regolare, anche se in verità ciò accade raramente. Questo potrebbe sembrare apparentemente in contrasto con i provati effetti positivi della terapia da desferrioxamina, ma è importare ricordare che i valori di ferritina non sono un’indicazione assoluta della quantità di ferro nel corpo, che possono essere influenzati anche da altri fattori, in particolare dalle malattie del fegato.
L’unico metodo realmente affidabile per misurare un eventuale accumulo di ferro è la biopsia del fegato, un esame purtroppo molto invasivo.
Gravidanza ed anemia mediterranea
Anche se nelle donne affette da talassemia dipendente da trasfusione la fertilità è ridotta, in certi casi non è da escludere la possibilità di una gravidanza naturale. In letteratura si rileva nn consistente numero di gravidanze in donne malate la maggior parte delle quali affette da beta talassemia intermedia ed una piccola parte da beta talassemia major.
Grazie alla disponibilità di tecniche di riproduzione assistita e grazie ad i continui progressi in campo medico, aumenterà parallelamente anche la qualità e la speranza di vita delle donne con beta talassemia, in più continuerà a crescere il numero di gravidanze portate a termine con successo.
Le domande più frequenti
Cos'è l'anemia mediterranea?
Quali sono i sintomi dell'anemia mediterranea?
Cosa significa portatore sano?
Come capire se si è affetti da anemia mediterranea?
Posso assumere ferro essendo portatrice sana di anemia mediterranea?
Cosa devono sapere una coppia di portatori sani di anemia mediterranea prima di avere figli?
Ci sono effetti collaterali nel prendere integratori di ferro per chi è portatore di anemia mediterranea?
Tutti gli aggiornamenti su salute, alimentazione e benessere.






