Introduzione
La poliarterite nodosa (PAN), conosciuta anche come malattia di Kussmaul-Maier, è una
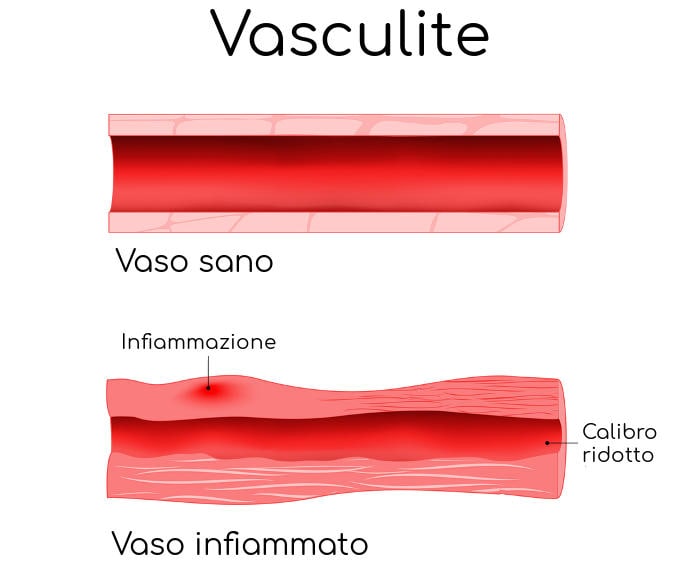
- vasculite (una patologia che prevede l’infiammazione dei vasi sanguini),
- necrotizzante (con possibilità che si associ la necrosi della parete vascolare, ossia la completa perdita di funzionalità),
- sistemica (che può colpire l’intero organismo).
Nella PAN il processo infiammatorio interessa le arterie di medio e piccolo calibro e più raramente arteriole o venule. Essendo sistemica la patologia può colpire qualsiasi organo. Un elemento distintivo fondamentale, che la differenzia da altre forme simili, è che la poliarterite nodosa non colpisce solitamente i piccoli vasi (capillari) e non è associata agli anticorpi ANCA (anticorpi anti-citoplasma dei neutrofili).
iStock.com/ttsz
La poliarterite nodosa è una malattia rara con incidenza di circa 1 caso ogni 100.000 abitanti, che si presenta soprattutto in età adulta (40-60 anni) e con frequenza ancora inferiore in età infantile.
Si tratta di una malattia idiopatica con cause ancora sconosciute, ma che si associa ad alcune condizioni come
- l’infezione da virus epatitici (epatite B e C),
- malattie autoimmuni,
- o sindromi mielodisplastiche.
Essendo una malattia sistemica i sintomi sono eterogenei e possono coinvolgere diversi organi ed apparati:
- sintomi generali (astenia, dolori muscolari ed articolari, febbre, calo ponderale),
- apparato gastrointestinale,
- cuore,
- reni,
- cute,
- sistema nervoso.
La diagnosi clinica prevede il riconoscimento di alcuni sintomi tipici associati ad alterazioni negli esami di laboratorio o reperti angiografici patologici; la conferma richiede sempre una biopsia tessutale di un organo coinvolto.
Il trattamento prevede l’utilizzo di farmaci cortisonici associati ad immunosoppressori nei casi più gravi. Questa terapia permette di migliorare notevolmente la prognosi dei pazienti. In caso di mancato trattamento la prognosi risulta infausta.
Cause
Nella maggior parte dei casi l’eziologia della PAN rimane sconosciuta, ovvero non si riesce a riconoscere una causa sottostante, tuttavia è stata rilevata un’associazione significativa tra la poliarterite nodosa e alcune condizioni patologiche come:
- infezione da HBV (virus dell’epatite B),
- infezione da HCV (virus dell’epatite C),
- tricoleucemia (leucemia cellule capellute),
- consumo di anfetamine,
- sindromi mielodisplastiche,
- malattie autoimmuni (artrite reumatoide, sindrome di Sjogren, Lupus, …).
I vasi coinvolti dalla poliarterite nodosa sono soprattutto le arterie di medio calibro che vanno incontro a necrosi della loro parete. Le lesioni hanno una distribuzione segmentaria con predilezione per le zone di biforcazione dei vasi.
Altra lesione frequente è la dilatazione aneurismatica (formazione di piccoli aneurismi) da sfiancamento della parete vasale infiammata, il cui rischio è quello di una rottura del vaso con conseguente emorragia.
La poliarterite nodosa è una vasculite sistemica che quindi può interessare qualsiasi organo, tuttavia in oltre il 70% dei pazienti vengono coinvolti i reni ed il cuore; molto raramente invece vengono interessati i polmoni o la milza.
Sintomi
In base al territorio vascolare interessato la poliarterite nodosa presenta una diversa sintomatologia, ma a prescindere da questo sono tipicamente presenti:
- febbre,
- astenia e malessere generale,
- artro-mialgia (dolori articolari e muscolari),
- riduzione dell’appetito,
- dimagrimento,
- lesioni a livello cutaneo.
Manifestazioni renali
Si presentano in oltre il 60% dei pazienti e sono correlate a fenomeni di ischemia renale. I principali segni di compromissione renale sono:
- proteinuria (perdita di proteine con le urine),
- microematuria (perdita di sangue con le urine non visibile ad occhio nudo),
- sindrome nefrosica,
- ipertensione nefrovascolare (sviluppo di pressione alta causata da una patologia renale)
- insufficienza renale acuta o cronica,
- rottura di aneurismi di vasi renali con ematomi perirenali.
Manifestazioni neurologiche
La poliarterite nodosa spesso esordisce con una neuropatia periferica, ovvero con lesioni a danno dei nervi periferici. La neuropatia prevede un dolore acuto e parestesia (alterazioni della sensibilità) nel territorio di pertinenza del nervo colpito. Il dolore e la parestesia vengono poi seguiti dopo alcune ore o pochi giorni da deficit motori anche gravi (paresi o paralisi).
Il danno nervoso può coinvolgere anche altri nervi ed evolvere verso un quadro di polineurite sensitiva e motoria.
Il coinvolgimento del sistema nervoso centrale (SNC) è meno frequente e può presentarsi con sintomi come:
- cefalea,
- TIA,
- ictus ischemico,
- emorragia cerebrale,
- crisi epilettica,
- disfunzione cerebellare,
- deficit motori,
- interessamento dei nervi cranici.
Manifestazioni articolari e muscolari
Quelli a carico di articolazioni e muscoli sono sintomi piuttosto frequenti. I dolori articolari insorgono tipicamente al mattino e spesso evolvono verso un quadro di poliartrite simmetrica non deformante che coinvolge le grandi articolazioni delle estremità di arti superiori ed inferiori (gomito, polso, mano, caviglia).
Il coinvolgimento dei vasi che irrorano i muscoli scheletrici porta a dolori muscolari piuttosto diffusi; spesso compare un dolore crampiforme notturno ai polpacci. Più raramente si presenta claudicatio intermittens (dolore alle gambe che compare dopo aver percorso brevi distanze di 100-200 metri).
Manifestazioni gastro-intestinali
Possono presentarsi in un 50% dei casi e prevedono:
- dolore addominale intermittente o continuo,
- angina abdominis (dolore addominale dopo aver mangiato),
- emorragie gastrointestinali:
- ematemesi (emissione di sangue dalla bocca),
- melena (emissioni di feci scure frammiste a sangue ossidato),
- vasculite di organi addominali (colecistite, appendicite, …).
Altri sintomi
A livello cardiaco è possibile rilevare:
- pericardite,
- insufficienza cardiaca,
- angina pectoris (dolore toracico),
- ipertensione arteriosa (pressione alta),
- infarto cardiaco se la vasculite coinvolge le coronarie.
A livello genito-urinario possono comparire:
A livello oculare il coinvolgimento di vasi retinici può portare a:
- emorragie o essudati retinici,
- disturbi della vista,
- dolore oculare,
- distacco di retina.
A livello cutaneo si presentano sintomi in oltre il 50% dei casi:
- livedo reticularis (alterazione del colore della pelle),
- porpora palpabile,
- noduli sottocutanei,
- lesioni ulcerative,
- alterazioni ischemiche con rischio ulcerativo alle estremità degli arti inferiori (quadro molto simile al “piede diabetico”).
Diagnosi
Il percorso diagnostico per la poliarterite nodosa è complesso a causa della natura multisistemica della malattia e della sua rarità. Non esiste un singolo test definitivo; la diagnosi si basa sulla combinazione di segni clinici, esami di laboratorio, tecniche di imaging e, idealmente, la conferma istologica.
Esami di laboratorio
Le analisi del sangue servono a valutare il grado di infiammazione e la funzionalità degli organi coinvolti:
- Indici di flogosi: si osserva quasi sempre un innalzamento della VES e della Proteina C-reattiva (PCR), insieme a un aumento dei globuli bianchi (leucocitosi) e delle piastrine.
- Screening anticorpale: un passaggio cruciale è la ricerca degli ANCA. Nella PAN classica, questi anticorpi sono tipicamente assenti (risultato negativo), aiutando a distinguerla da altre vasculiti come la poliangioite microscopica.
- Test virali: è fondamentale ricercare la presenza di infezioni da virus dell’epatite B (HBV) e C (HCV), poiché il trattamento differisce significativamente nelle forme virus-correlate.
- Analisi delle urine: si ricercano segni di danno renale, come la presenza di proteine o microematuria.
Imaging radiologico
Quando il sospetto clinico è orientato verso la PAN, lo studio dei vasi è prioritario:
- Angiografia: rimane un esame di riferimento. Mostra tipicamente piccoli aneurismi (dilatazioni) alternati a restringimenti (stenosi) dei vasi arteriosi, conferendo il caratteristico aspetto “a corona di rosario”.
- Angio-TC e Angio-RM: queste tecniche meno invasive sono oggi ampiamente utilizzate per visualizzare le infiammazioni dei vasi addominali, renali e cerebrali, permettendo di identificare aneurismi o occlusioni.
Biopsia
La conferma definitiva richiede l’analisi al microscopio di un frammento di tessuto (biopsia). Il medico sceglie il sito del prelievo in base ai sintomi: una zona cutanea con noduli o ulcere, un nervo periferico se è presente una neuropatia, o il tessuto muscolare. L’esame istologico deve mostrare una vasculite necrotizzante delle arterie di medio o piccolo calibro, senza evidenza di granulomi.
Criteri di classificazione
Per facilitare il riconoscimento, i medici utilizzano criteri clinici consolidati. La diagnosi è probabile se sono presenti almeno tre dei seguenti elementi: perdita di peso superiore a 4 kg, livedo reticularis, dolore testicolare, mialgie o debolezza muscolare, neuropatia, ipertensione di nuova insorgenza, alterazioni dei parametri renali, positività per l’epatite B, o reperti angiografici/bioptici caratteristici.
Cura
L’obiettivo primario della terapia è spegnere rapidamente l’infiammazione vascolare per prevenire danni irreversibili agli organi (come l’infarto renale o intestinale) e indurre una remissione duratura della malattia. Il piano terapeutico è strettamente personalizzato in base alla gravità del quadro clinico e all’eventuale associazione con infezioni virali.
Approccio farmacologico standard
Nelle forme idiopatiche (senza causa nota), il trattamento si divide in due fasi:
- Fase di induzione: mira a controllare rapidamente la malattia. Prevede l’uso di farmaci corticosteroidei ad alte dosi (spesso iniziati per via endovenosa). Nelle forme gravi che colpiscono organi vitali o il sistema nervoso, al cortisone si associa un potente immunosoppressore, solitamente la ciclofosfamide. In casi selezionati e refrattari, possono essere impiegati farmaci biologici come il rituximab.
- Fase di mantenimento: una volta ottenuta la remissione (solitamente dopo 3-6 mesi), si passa a farmaci con minor profilo di tossicità a lungo termine, come l’azatioprina o il metotrexato, riducendo progressivamente il dosaggio del cortisone.
Trattamento della PAN associata a Epatite B
Se la vasculite è scatenata dal virus HBV, l’approccio è differente. L’obiettivo è eliminare il virus e i complessi immunitari circolanti. La terapia prevede:
- Farmaci antivirali: specifici per l’epatite B.
- Plasmaferesi (scambio plasmatico): una procedura che “pulisce” il sangue dagli anticorpi e dai complessi infiammatori.
- Uso limitato di immunosoppressori: il cortisone viene usato solo per brevi periodi per evitare di favorire la replicazione virale.
Gestione delle complicanze e stile di vita
Il supporto al paziente non si limita ai soli farmaci immunosoppressori:
- Controllo pressorio: l’ipertensione è comune e va trattata con rigore per proteggere i reni e il cuore.
- Stile di vita: è fondamentale la cessazione totale del fumo, poiché il tabagismo aggrava ulteriormente il danno vascolare. Una dieta povera di sale aiuta a gestire la pressione arteriosa e gli effetti collaterali del cortisone.
- Riabilitazione: in caso di danni neurologici o motori, la fisioterapia è essenziale per il recupero funzionale degli arti.
- Monitoraggio costante: il paziente deve sottoporsi a controlli periodici della funzionalità renale, cardiaca e degli indici di infiammazione per intercettare precocemente eventuali ricadute.
Con i moderni protocolli terapeutici, la sopravvivenza a 5 anni supera l’80%, trasformando quella che un tempo era una malattia quasi sempre fatale in una condizione cronica gestibile nella maggior parte dei casi.
Fonti e bibliografia
- “Malattie dei reni e delle vie urinarie” – F.P. Schena, F.P. Selvaggi, L. Gesualdo, M. Battaglia. Ed. McGraw-Hill – quarta edizione
- EULAR recommendations for the management of systemic vasculitis.
- ACR/EULAR classification criteria for polyarteritis nodosa.
Tutti gli aggiornamenti su salute, alimentazione e benessere.






