Cos’è l’amiloidosi (in parole semplici)?
L’amiloidosi è una malattia rara che si verifica quando alcune proteine, che normalmente circolano nel sangue, iniziano a comportarsi in modo anomalo: invece di mantenere la loro forma corretta, si ripiegano male e si aggregano tra loro, formando dei depositi fibrosi (chiamati amiloidi) che si accumulano in vari organi.
È come se questi depositi “impregnassero” i tessuti, impedendo agli organi di funzionare correttamente.
Ne esistono diverse forme (oltre 40) e purtroppo ad oggi non esiste una cura definitiva.
Amiloidosi da transtiretina
Una forma particolare di amiloidosi è quella causata dalla proteina transtiretina (chiamata anche TTR) nella sua forma naturale, detta “wild type”.
Questa forma, purtroppo nota per aver causato la morte del fotografo Oliviero Toscani nel gennaio del 2025, colpisce principalmente le persone anziane, soprattutto uomini sopra i 65 anni.
La transtiretina è una proteina prodotta dal fegato che normalmente trasporta ormoni tiroidei e vitamina A nel sangue.
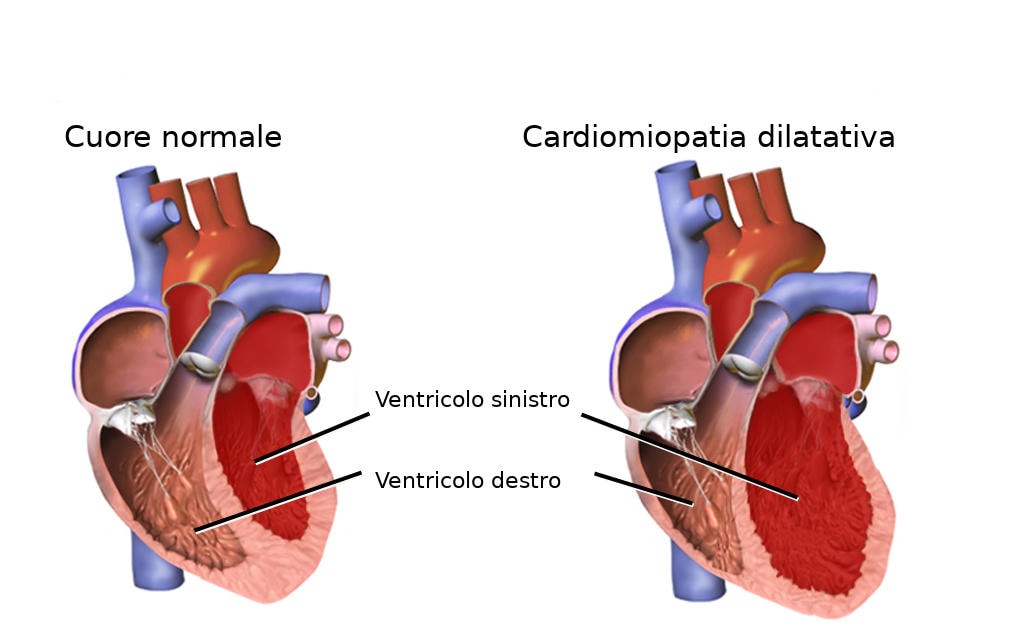
Con l’avanzare dell’età, nei pazienti colpiti dalla malattia, questa proteina diventa instabile: è come se i “mattoncini” che la compongono si staccassero e si riattaccassero in modo disordinato, formando delle fibrille che si depositano principalmente nel cuore.
I pazienti affetti da amiloidosi cardiaca possono sviluppare tra l’altro disturbi come
- un battito cardiaco anomalo (aritmia),
- un cuore ingrossato (cardiomegalia)
- e ipertensione ortostatica.
Questo accumulo progressivo rende il muscolo cardiaco più rigido e meno efficiente nel suo lavoro di pompa (insufficienza cardiaca). Poiché questa forma di amiloidosi è legata all’invecchiamento naturale della proteina (e non a mutazioni genetiche), viene chiamata “wild type” o “senile”.
Cause
Esistono diverse forme di amiloidosi, che dipendono dal tipo di proteina depositata e da quali tessuti sono coinvolti; nel 90% dei casi i depositi di fibrille sono formati da proteina amiloide dalla struttura alterata che si trasforma in aggregati insolubili per poi accumularsi, mentre nel 10% sono costituiti da altre molecole (ad esempio cheratina, calcitonina e fattore natriuretico atriale).
Si pensa che le forme localizzate della malattia siano causate da una produzione locale delle proteine responsabili della precipitazione, più che da una deposizione di quelle presenti nel sangue; molti meccanismi contribuiscono alla precipitazione degli aggregati proteici nei tessuti e quindi alla genesi della malattia, come:
- patologica degradazione delle proteine (proteolisi),
- proteolisi deficitaria o assente,
- mutazioni genetiche che provocano alterazioni delle proprietà termodinamiche e di trasporto delle proteine,
- infezioni croniche,
- altri meccanismi non ancora pienamente definiti.
Classificazione
Esistono circa trenta diversi tipi di amiloidosi, in base alla forma, la funzione, la sequenza di amminoacidi e la struttura delle fibrille coinvolte.
Le più frequenti amiloidosi sono:
- Amiloidosi da catene leggere (AL – amyloid light chains). Anche chiamata amiloidosi primaria, è il tipo più comune ed è causata dall’accumulo di immunoglobuline (anticorpi) formate da catene leggere. Anche se la causa esatta non è nota, questo tipo insorge quando il midollo osseo produce un numero elevato di anticorpi che non possono essere smaltiti. Questa condizione è associata al mieloma multiplo, un tipo di tumore che colpisce le cellule del sangue. Le catene leggere si depositano nei reni, cuore, fegato, intestino e neuroni.
- Amiloidosi reattiva a infiammazione cronica (AA – amyloid type A protein). In passato denominata amiloidosi secondaria, è causata da alcune condizioni infiammatorie croniche come l’artrite reumatoide, il morbo di Crohn o la rettocolite ulcerosa. Colpisce principalmente i reni, ma può interessare anche l’apparato digerente, il fegato e il cuore.
- Amiloidosi correlata a dialisi (DRA – dialysis related amyloidosis). A causa della dialisi è il tipo più comune nelle persone anziane o che sono state in dialisi per più di 5 anni. La proteina coinvolta è la beta2-microglobulina e si deposita più frequentemente a livello delle ossa, articolazioni e tendini.
- Amiloidosi familiari o ereditarie (ATTR, AAPOAI, ecc). Queste forme sono condizioni rare trasmesse per via ereditaria che spesso coinvolgono fegato, neuroni, cuore e reni. Esistono diverse mutazioni genetiche che possono predisporre allo sviluppo della malattia, ad esempio la mutazione della proteina transtiretina (TTR) o dell’apolipoproteina AI (APOAI).
- Amiloidosi senile. È determinata dall’accumulo della proteina TTR mutata a livello neuronale, del cuore e in altri tessuti. È più frequente nel sesso maschile.
- Amiloidosi organo-specifica. Si tratta di una forma che si verifica quando le proteine si accumulano nei singoli organi, come ad esempio l’amiloidosi cutanea da accumulo di cheratina, il carcinoma midollare della tiroide dovuto ad accumulo di calcitonina, o l’amiloidosi atriale dovuta all’accumulo di fattore natriuretico atriale a livello cardiaco.
Fattori di rischio
I fattori di rischio per lo sviluppo della malattia sono rappresentati da:
- sesso maschile (nel 70% dei casi di amiloidosi AL),
- età compresa fra i 60 e 70 anni,
- presenza di malattie concomitanti come le infezioni croniche (per l’amiloidosi AA) o il mieloma multiplo (per l’amiloidosi AL),
- presenza di insufficienza renale cronica e dialisi (amiloidosi correlata a dialisi DRA).
Sintomi

Shutterstock/PeopleImages.com – Yuri A
I sintomi dell’amiloidosi sono estremamente variabili in base ai tessuti colpiti, ma le manifestazioni sistemiche più comuni sono relative al coinvolgimento di cuore e rene:
- aritmie (irregolarità del battito cardiaco),
- dispnea (difficoltà di respirazione),
- fatica,
- gonfiore delle caviglie (edema),
- anemia,
- ritenzione idrica,
- perdita di proteine nelle urine (proteinuria).
Tra gli altri possibili sintomi ricordiamo invece:
- variazione del colore della pelle,
- decadimento cognitivo,
- sensazione di pienezza,
- dolore articolare,
- rigonfiamento della lingua,
- difficoltà nella deglutizione,
- formicolio e intorpidimento di gambe e piedi,
- debolezza nelle mani,
- perdita di peso improvvisa.
Amiloidosi cardiaca
Il deposito di fibrille a livello cardiaco può causare un irrigidimento delle pareti cardiache e una diminuzione della funzionalità e dell’attività elettrica del cuore.
In situazioni gravi potrebbe verificarsi insufficienza cardiaca perché i muscoli della parete non sono più in grado di pompare il sangue in maniera adeguata.
I sintomi caratteristici sono
- aritmie;
- segni di scompenso cardiaco:
- caviglie e piedi gonfi (edema),
- debolezza,
- fatica,
- dispnea,
- nausea,
- …
Amiloidosi renale
Quando le proteine si depositano a livello del rene, possono comprometterne la funzione di filtro. Il rene, infatti, elimina le sostanze tossiche e l’acqua in eccesso e, se questa funzione è interrotta, si accumulano nell’organismo provocando:
- segni di insufficienza renale:
- aumento della creatinina nel sangue,
- ritenzione idrica che si manifesta con
- una diminuzione della produzione di urina,
- edemi alle caviglie, ai piedi e intorno agli occhi;
- perdita nelle urine delle proteine importanti per il corpo (proteinuria);
Amiloidosi gastro-intestinale
I depositi di amiloide a livello intestinale interferiscono con le funzioni di trasporto, digestione e assimilazione del cibo portando a:
- perdita dell’appetito,
- diarrea,
- nausea,
- gastralgia (dolore allo stomaco),
- perdita di peso,
- epatomegalia (ingrossamento del fegato).
Neuropatia amiloide (amiloidosi cerebrale)
Quando l’amiloidosi colpisce il cervello, i nervi periferici e il midollo spinale, le informazioni raccolte dall’ambiente esterno non sono integrate a livello cerebrale. Si manifesteranno quindi problemi di
- equilibrio,
- controllo della vescica e dell’intestino,
- sudorazione,
- formicolio e stanchezza a gambe e braccia,
- ipotensione (pressione bassa) e svenimenti,
- decadimento cognitivo.
Complicazioni
Negli ultimi anni sono stati fatti straordinari progressi nella terapia delle amiloidosi e la prognosi dei pazienti è in continuo miglioramento.
Possono presentarsi comunque alcune complicazioni anche gravi:
- Renali:
- proteinuria (perdita di proteine nelle urine),
- ritenzione idrica e di sostanze tossiche fino all’insufficienza renale;
- Cardiache:
- aritmie (anomalie nella conduzione elettrica del cuore),
- ipertrofia delle pareti con irrigidimento e diminuzione della funzione di pompa fino allo scompenso cardiaco;
- Neuronali:
- dolore,
- intorpidimento e formicolio,
- mancanza di sensibilità e bruciore ai piedi o alle dita,
- alterazioni dell’alvo (alternanza di stitichezza e diarrea) se è coinvolta l’innervazione intestinale,
- ipotensione e svenimento se è colpita l’innervazione che controlla la pressione arteriosa,
- decadimento cognitivo.
Diagnosi
Un’accurata diagnosi è fondamentale per stabilire la terapia più adeguata, in quanto esistono terapie distinte secondo il meccanismo coinvolto; purtroppo a causa dei sintomi aspecifici e difficili da riconoscere il ritardo nella diagnosi è ancora un evento comune.
L’iter diagnostico in genere prevede:
- raccolta dei dati personali e familiari (anamnesi patologica),
- esame fisico accurato,
- esami di laboratorio ematici e delle urine,
- biopsia con agoaspirato ed esame istologico per confermare la diagnosi e identificare l’esatta proteina responsabile,
- esami strumentali per quantificare il danno d’organo:
- analisi genetiche qualora il medico sospetti una forma ereditaria di amiloidosi.
Non è sempre necessaria la biopsia dell’organo danneggiato, poiché i capillari sottocutanei sono spesso interessati dal processo di deposito e sono quindi sufficienti al fine della diagnosi.
Il campione bioptico più spesso utilizzato è quello del tessuto adiposo addominale periombelicale; si tratta di un prelievo di un piccolo campione di grasso vicino all’ombelico, eseguito attraverso un ago sottile e in seguito a leggera anestesia locale. Il campione viene poi colorato con appositi coloranti e illuminato al microscopio con speciali filtri. In presenza dei depositi di amiloide il tessuto prelevato da rosso diventa verde.
Se la biopsia del grasso addominale non conduce alla diagnosi, ma il medico sospetta ancora la malattia, si può optare per una biopsia di un diverso campione, come ad esempio:
- ghiandola salivare,
- mucosa rettale,
- midollo osseo,
- cavità orale,
- o di altri organi.
Cura
L’obiettivo terapeutico nell’amiloidosi è rallentare o impedire la formazione delle fibrille amiloidi e, quando possibile, favorire la dissoluzione dei depositi proteici già formati. Le strategie terapeutiche variano significativamente in base al tipo di amiloidosi, alla proteina coinvolta e agli organi interessati.
Nel caso di coinvolgimento del cuore potrebbe essere necessario il ricorso alla somministrazione di anticoagulanti per ridurre il rischio di coaguli ed eventualmente medicinali per controllare la frequenza cardiaca e diuretici per ridurre lo sforzo sui reni e sullo stesso muscolo cardiaco.
Nell’amiloidosi da transtiretina (ATTR), sia wild-type che ereditaria, disponiamo oggi di farmaci specifici che agiscono attraverso due meccanismi principali:
- gli stabilizzatori della TTR (come tafamidis e diflunisal) che prevengono la dissociazione della proteina
- e i silenziatori genici (patisiran e inotersen) che riducono la produzione epatica di TTR.
Per l’amiloidosi AL, associata alla produzione anomala di catene leggere delle immunoglobuline, il trattamento si è evoluto significativamente negli ultimi anni. Oltre alla tradizionale chemioterapia ad alte dosi seguita dal trapianto autologo di cellule staminali, disponiamo ora di un arsenale terapeutico che include inibitori del proteasoma (bortezomib, carfilzomib), immunomodulatori (lenalidomide) e anticorpi monoclonali (daratumumab). La scelta del trattamento viene personalizzata in base alle caratteristiche del paziente e alla gravità della malattia.
Nell’amiloidosi AA, secondaria a infiammazione cronica, l’approccio terapeutico moderno si concentra sul controllo della patologia infiammatoria di base attraverso terapie biologiche mirate, come gli inibitori del TNF-α, dell’interleuchina-1 e dell’interleuchina-6. Questo approccio ha largamente sostituito l’uso generico di farmaci antiinfiammatori.
Il trapianto d’organo mantiene un ruolo importante in casi selezionati: il trapianto di fegato può essere indicato in alcune varianti di ATTR ereditaria, mentre il trapianto renale viene considerato come opzione terapeutica nello stadio terminale della malattia renale (dopo un’eventuale transizione con dialisi), non come trattamento della patologia di base.
La possibilità di recupero funzionale degli organi colpiti dipende da diversi fattori: il tipo specifico di amiloidosi, gli organi coinvolti, lo stadio della malattia al momento della diagnosi e la tempestività del trattamento. In alcuni casi, se il danno non è diventato irreversibile, la funzione degli organi può essere recuperata anche in modo significativo.
Parallelamente alla terapia specifica, è fondamentale una gestione multidisciplinare che includa terapie di supporto mirate agli organi interessati: supporto cardiologico, nefrologico e neurologico, terapia del dolore, supporto nutrizionale e psicologico, programmi di riabilitazione fisica e gestione delle complicanze autonomiche. Particolare attenzione viene dedicata alla prevenzione delle cadute nei pazienti con neuropatia. Questo approccio integrato è essenziale per migliorare la qualità della vita del paziente e ottimizzare i risultati del trattamento.