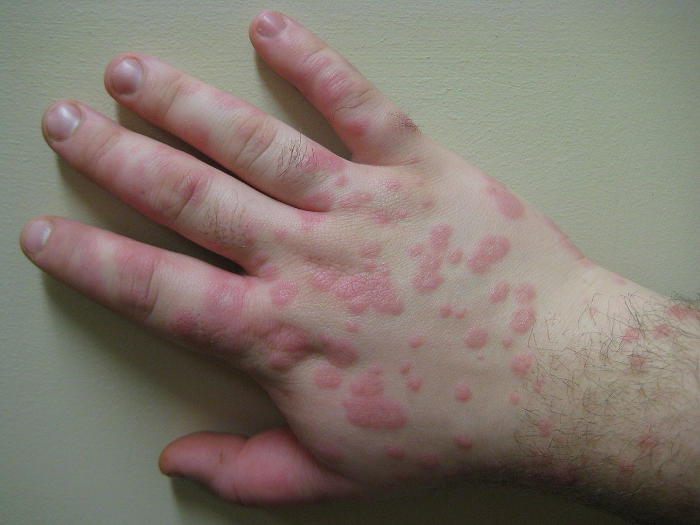
Introduzione
La sindrome di Stevens-Johnson (SSJ) è una grave reazione cutanea che, il più delle volte, viene innescata da specifici farmaci; sebbene in passato si pensasse che sindrome di Stevens-Johnson e la necrolisi epidermica tossica fossero condizioni separate, sono ad oggi considerate parte di un continuum, in cui la sindrome di Stevens-Johnson rappresenta l’estremità meno grave dello spettro della malattia e la necrolisi epidermica tossica la forma più grave.
In altre parole possono essere considerate due forme delle stesso disturbo cutaneo, peraltro potenzialmente letale, differenziate in base all’estensione del danno alle pelle:
- Sindrome di Stevens-Johnson: meno del 10% della superficie corporea
- Sovrapposizione sindrome di Stevens-Johnson/necrolisi epidermica tossica: dal 10% al 30% della superficie corporea
- Necrolisi epidermica tossica superiore al 30% della superficie corporea.
La condizione esordisce in genere con lo sviluppo di febbre e sintomi simil-influenzali, ma che nel giro di pochi giorni si manifesta in tutta la sua gravità, con la formazione di vesciche e processi desquamativi della pelle. Si osservano aree grezze molto dolorose (erosioni) che hanno l’aspetto di una grave ustione da acqua calda.
Le erosioni cutanee compaiono inizialmente sul viso e sul torace, per poi diffondersi ad altre parti del corpo. Nella maggior parte delle persone colpite, la condizione interessa anche le mucose, compreso il rivestimento della bocca e le vie aeree, con il rischio di sviluppare difficoltà di deglutizione e respirazione. Le dolorose vesciche possono interessare anche le vie urinarie e i genitali, oltre che gli occhi, dove causano irritazione e arrossamento della congiuntiva.
I danni sono così severi da costituire un pericolo per la vita del paziente colpito; poiché la pelle normalmente funge da barriera protettiva, un danno cutaneo così esteso può rapidamente condurre a una pericolosa perdita di liquidi e favorire lo sviluppo di infezioni.
La sindrome di Stevens-Johnson è un’emergenza medica che richiede il ricovero in ospedale.
La terapia prevede ovviamente la rimozione della causa (ad esempio la sospensione del farmaco responsabile), la cura delle piaghe, il controllo del dolore e la prevenzione delle possibili complicazioni.
Richiede spesso settimane, talvolta mesi per un completo recupero.
Cause
La sindrome di Stevens-Johnson è una condizione rara, che interessa meno di 10 pazienti ogni milione di persone all’anno.
Si tratta di una reazione del corpo che è tipicamente innescata dall’assunzione di determinati medicinali (4 pazienti su 5), come ad esempio:
- medicinali contro l’epilessia
- carbamazepina
- lamotrigina
- fenobarbitale
- fenitoina
- antibiotici e antiretrovirali
- sulfametossazolo e altri antibiotici sulfamidici
- nevirapina
- antidolorifici antinfiammatori
- meloxicam
- piroxicam
- sulfasalazina
- e talvolta anche molecole più comuni come paracetamolo, ibuprofene, naprossene, …
- allopurinolo (farmaco usato contro la gotta).
Nei bambini, la sindrome di Stevens-Johnson viene talvolta scatenata da infezioni virali come
Nell’adulto sono stati segnalati casi associati a
- polmonite da Mycoplasma pneumoniae,
- infezioni virali in genere, tra cui il citomegalovirus.
Fattori di rischio
Pazienti affetti da tumori del sangue, sieropositivi per HIV o affetti da lupus eritematoso sistemico hanno maggiori probabilità di sviluppare la sindrome rispetto alla popolazione generale, seppure la ragione non sia ancora stata chiarita (l’ipotesi prevalente verte ovviamente su possibili alterazioni del sistema immunitario).
Esistono poi alcune predisposizioni genetiche allo sviluppo della malattia e la maggior parte di queste alterazioni è localizzata nei geni coinvolti nella normale funzione del sistema immunitario.
Le variazioni genetiche più fortemente associate alla sindrome di Stevens-Johnson si verificano nel gene HLA-B; si tratta di un gene che fa parte di una famiglia chiamata complesso dell’antigene leucocitario umano (HLA). Il complesso HLA aiuta il sistema immunitario a distinguere le proteine del corpo da quelle prodotte da invasori estranei (come virus e batteri).
Sintomi
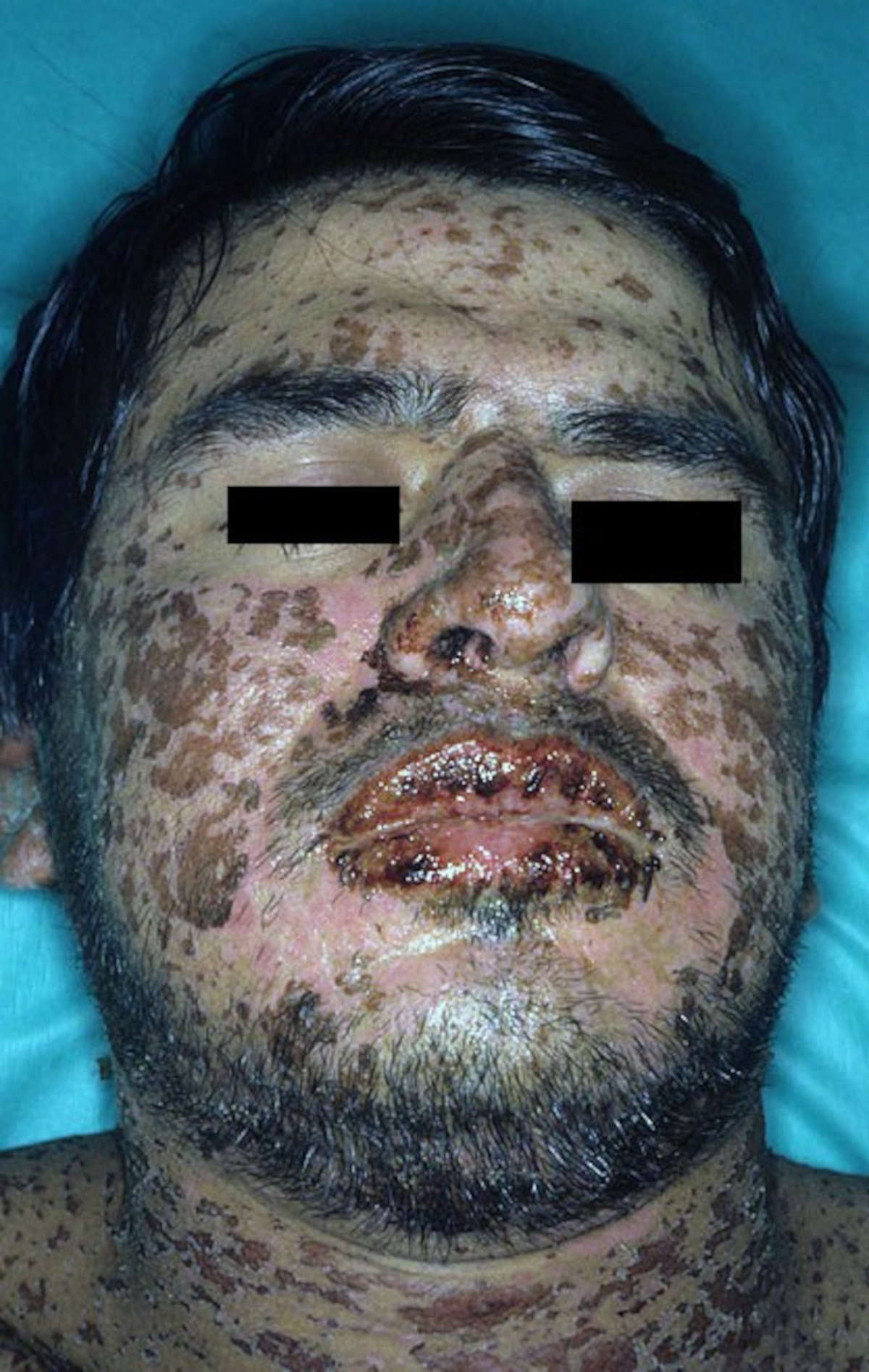
By Dr. Thomas Habif – http://www.dermnet.com/Stevens-Johnson-Syndrome/picture/14975, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=9699327
La sindrome di Stevens-Johnson può iniziare con sintomi simil-influenzali, come febbre alta, mal di gola, tosse e dolori articolari.
Dopo circa 3-4 giorni appare l’eruzione cutanea, caratterizzata dallo sviluppo di dolorose vesciche, che di solito inizia nella parte superiore del corpo, per poi diffondersi rapidamente al viso, alle braccia, alle gambe e ad altre aree del corpo, compresi i genitali.
In genere NON è accompagnata da prurito, a differenza del dolore (peraltro molto severo).
Purtroppo le vesciche possono comparire anche a livello di:
- labbra, bocca e in gola, fino a rendere dolorosa la deglutizione
- sistema urinario, rendendo dolorosa la minzione
- occhi, con un dolore peggiorato dalla luce (fotofobia) e lo sviluppo di problemi di vista.
Le vesciche guariscono in 2 o 3 settimane a meno di complicazioni (infezioni), che possono allungare i tempi.
Da un punto di vista generale il paziente si presenta molto provato,ansioso e dolorante. Anche gli organi interni possono essere interessati dalla sindrome (fegato, reni, polmoni, midollo osseo e articolazioni) con lo sviluppo di:
- malessere, mal di testa, anoressia
- disturbi alla vista, polmonari, cardiovascolari, gastrointestinali, renali ed ematologici a seconda degli apparati/sistemi che vengono coinvolti.
Complicazioni
Il recupero dalla sindrome di Stevens-Johnson richiede tempi lunghi, settimane o mesi, durante il quale il paziente avverte una profonda stanchezza.
Le possibili complicanze possono includere
- disidratazione,
- polmonite,
- infezioni batteriche sistemiche (sepsi),
- shock,
- insufficienza multi-organo
- e morte (10% dei pazienti colpiti da sindrome di Stevens-Johnson e 50% di quelli con necrolisi epidermica tossica).
Tra le persone che sopravvivono, gli effetti a lungo termine della sindrome di Stevens-Johnson annoverano:
- alterazioni del colore della pelle e cicatrici,
- secchezza della pelle e delle mucose (xerosi),
- sudorazione eccessiva (iperidrosi),
- perdita di capelli (alopecia)
- e crescita anormale o perdita delle unghie delle mani e dei piedi.
Altri problemi a lungo termine possono includere alterazione del gusto, difficoltà a urinare e anomalie genitali.
Una piccola percentuale di individui sviluppa infine secchezza cronica o infiammazione degli occhi, che può portare a una maggiore sensibilità alla luce (fotofobia) e compromissione della vista.
Diagnosi
La diagnosi della sindrome di Stevens-Johnson è clinica e viene in genere formulata da un dermatologo o da un oculista, a seguito di un’approfondita visita medica; in casi dubbi è possibile procedere alla biopsia (prelievo di un frammento di pelle e successiva analisi di laboratorio) per la diagnosi di certezza.
Cura
La sindrome di Stevens-Johnson richiede il ricovero ospedaliero (spesso in terapia intensiva o nel reparto grandi ustionati) e la collaborazione di numerose figure specialistiche:
- Intensivista
- Dermatologo
- Specialista in chirurgia plastica o ustioni
- Oculista
- Ginecologo
- Urologo
- Pneumologo
- Fisioterapista
- Nutrizionista.
La terapia è molteplice e prevede:
- immediata sospensione del farmaco responsabile
- somministrazione di fluidi per endovena
- monitoraggio dei parametri vitali
- sollievo dal dolore
- ossigeno supplementare e, in alcuni casi, intubazione con ventilazione meccanica
- eventuale inizio dell’alimentazione con sondino nasogastrico
- trattamento topico delle aree interessate dalla presenza di vesciche, mediante impacchi freddi, creme e medicazioni avanzate (con particolare attenzione agli occhi)
- rimozione della pelle necrotica/tessuto mucoso
- eventuali coperture antibiotiche, solo al bisogno
- prevenzione e trattamento di altre complicazioni.
Benché l’evidenza di efficacia clinica sia limitata, spesso vengono prescritti a dosi elevate farmaci cortisonici per i primi tre-cinque giorni dal ricovero.
Prevenzione
A meno di spiccata familiarità è difficile tentare di prevedere l’insorgenza della condizione, mentre in caso di sviluppo della sindrome a seguito dell’assunzione di un farmaco questo andrà ovviamente evitato per il resto della vita del paziente.
Fonti e bibliografia
- NHS
- MedLinePlus
- Stevens-Johnson Syndrome – Amanda M. Oakley; Karthik Krishnamurthy
- John Hopkins
Tutti gli aggiornamenti su salute, alimentazione e benessere.