Introduzione
La malattia di Fabry (tecnicamente nota anche come carenza di alfa-galattosidasi-A) è una malattia neurologica ereditaria che si verifica quando l’enzima alfa-galattosidasi-A non è in grado di digerire in modo efficiente i grassi. Il difetto genetico causa un progressivo e pericoloso accumulo dei grassi a livello del sistema nervoso autonomo del corpo (la parte del sistema nervoso che controlla le funzioni involontarie come la respirazione e il battito cardiaco), così come negli occhi, nei reni e nel sistema cardiovascolare.
Ne sono più gravemente colpiti i maschi, anche se una forma più lieve può essere osservata anche nel sesso femminile. L’insorgenza dei sintomi avviene in genere durante l’infanzia o l’adolescenza, con lo sviluppo di caratteristici segni neurologici che includono:
- Bruciore alle braccia e alle gambe, che peggiora con il caldo o dopo l’esercizio fisico
- Accumulo di materiale in eccesso negli strati chiari della cornea (con conseguente vista annebbiata)
- Circolazione alterata che può favorire lo sviluppo di ictus e infarto a causa dell’accumulo di grasso nelle pareti dei vasi sanguigni.
Tra gli altri sintomi si annoverano:
- Ingrandimento patologico del cuore
- Insufficienza renale progressiva
- Disturbi gastrointestinali
- Riduzione della sudorazione
- Febbre
- Comparsa di angiocheratomi, piccoli punti in rilievo sulla pelle, non cancerosi e di colore rosso-violaceo, che possono svilupparsi nella parte inferiore del tronco del corpo e diventare più numerosi con l’età.
La terapia prevede la somministrazione dell’enzima mancante, consentendo di ridurre l’accumulo di lipidi, alleviare il dolore e preservare la funzione degli organi. Possono inoltre venire prescritti medicinale per trattare il dolore e il disagio gastrointestinale che accompagna la malattia di Fabry, ma che purtroppo non sono in grado di modificare l’andamento della malattia, così come principi attivi antiaggreganti e anticoagulanti per la prevenzione degli eventi cardiovascolari.
Cause
La malattia di Fabry è una malattia ereditaria che deriva “dall’accumulo di grasso (in particolare in forma di globotriaosilceramide) nei tessuti viscerali e nell’endotelio vascolare di tutto l’organismo, con danni a livello renale, cardiaco e del sistema nervoso centrale tali da compromettere qualità e aspettativa di vita”.
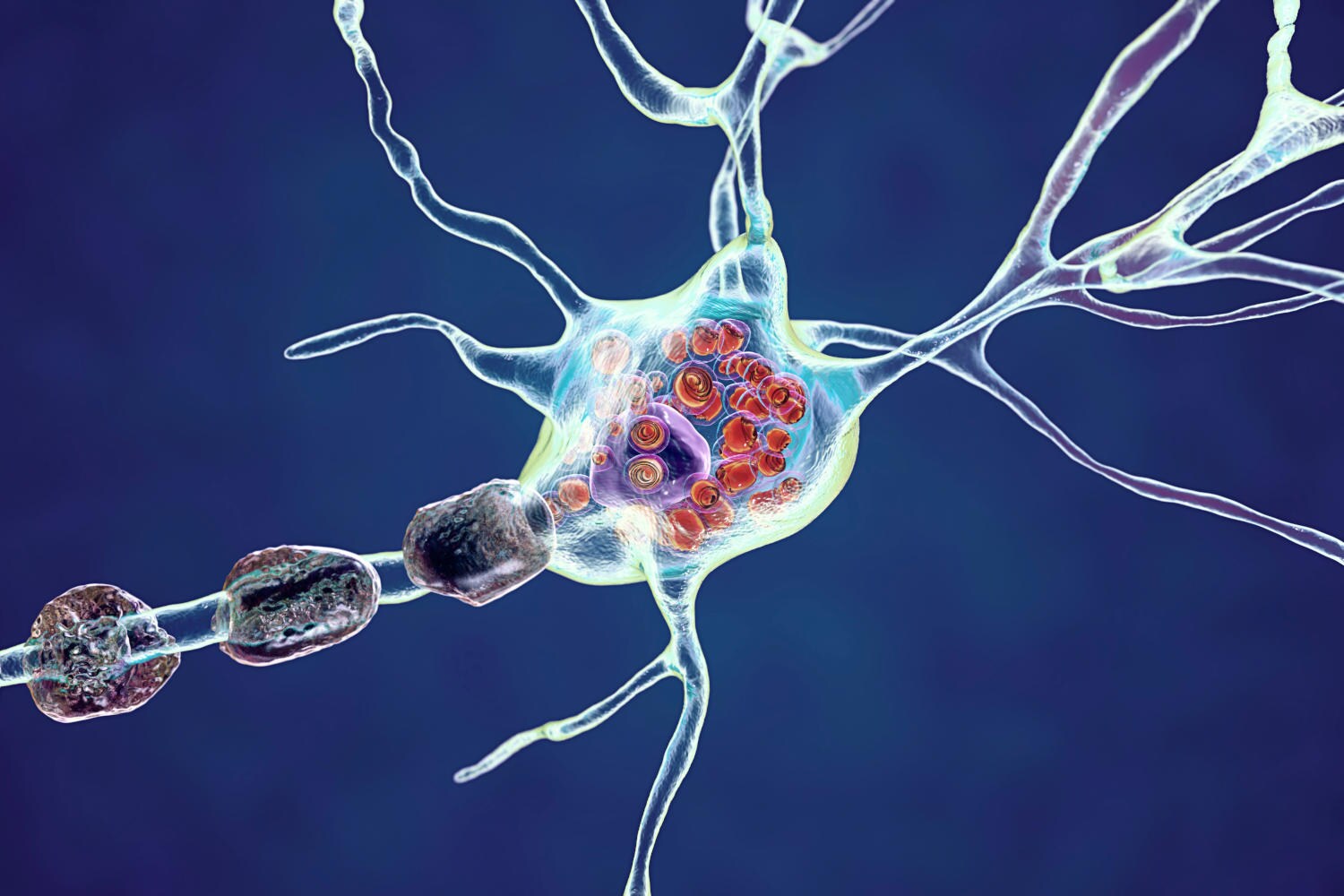
Shutterstock/Kateryna Kon
Colpisce un soggetto ogni 1.000-9.000 e le forme più lievi e tardive sembrano più comuni della più nota forma grave.
È causata da mutazioni genetiche, in particolare a livello del gene GLA, che contiene le istruzioni per produrre un enzima chiamato alfa-galattosidasi A. Questo enzima, attivo nei lisosomi (strutture che fungono da centri di riciclaggio all’interno delle cellule) è responsabile della scomposizione di una sostanza grassa chiamata globotriaosilceramide.
Le varianti difettose del gene GLA alterano la struttura e la funzione dell’enzima, impedendogli di scomporre efficacemente la sostanza, con la conseguenza di un pericoloso accumulo di globotriaosilceramide nelle cellule di tutto il corpo, in particolare quelle che rivestono i vasi sanguigni della pelle e nelle cellule di reni, cuore e sistema nervoso. Il progressivo accumulo di questa sostanza danneggia le cellule, manifestandosi con i diversi segni e sintomi della malattia di Fabry.
- Le varianti del gene GLA che determinano una completa incapacità di funzionare dell’enzima sono responsabili della forma grave,
- le varianti che riducono ma non impediscono del tutto l’attività dell’enzima di solito causano forme più lievi e tardive della malattia, che in genere colpiscono solo il cuore, i reni o i vasi sanguigni nel cervello.
Ereditarietà
Questa condizione viene trasmessa secondo il modello X-linked, ovvero “Ereditarietà legata al cromosoma X”.
Una condizione è considerata legata all’X se il gene alterato, causa del disturbo, si trova sul cromosoma X, uno dei due cromosomi sessuali presenti in ciascuna cellula:
- Nei maschi (che possiedono un solo cromosoma X) una copia alterata del gene GLA è sufficiente a causare la condizione;
- poiché le femmine hanno invece due copie del cromosoma X, una sola copia alterata del gene si manifesta con sintomi meno gravi, talvolta addirittura nessun sintomo, grazie ai meccanismi di compensazione possibili grazie al secondo cromosoma (se sano).
A differenza di altre malattie legate all’X la malattia di Fabry causa tuttavia complicazioni comunque significative in molte donne, nonostante una sola copia alterata del gene GLA; queste donne possono manifestare molte delle caratteristiche classiche del disturbo, tra cui anomalie del sistema nervoso, problemi ai reni, dolore cronico e affaticamento. Hanno anche un aumentato rischio di sviluppare pressione alta, malattie cardiache, ictus e insufficienza renale.
Sintomi
I maschi possono iniziare a manifestare i primi disturbi fin dall’infanzia e dalla prima adolescenza, con il possibile coinvolgimento di quasi tutti i sistemi/apparati; tra i sintomi più comuni figurano:
- acroparestesie dolorose (sensazione di intorpidimento e formicolio alle dita delle mani e dei piedi),
- riduzione della sudorazione e della lacrimazione,
- sintomi gastrointestinali, in particolare crampi addominali e diarrea,
- manifestazioni cutanee che includono piccole petecchie (lividi) attorno all’ombelico e soprattutto angiocheratomi (papule in rilievo di colore rosso, violaceo o bluastro)
- disturbi agli occhi e alla vista.
Altri sintomi che possono essere associati alla malattia di Fabry comprendono:
- stanchezza e affaticamento cronico,
- vertigini,
- mal di testa,
- debolezza generalizzata,
- nausea e/o vomito,
- pubertà ritardata (e mancanza o scarsa crescita dei peli).
Alcuni pazienti possono presentare ingrossamento dei linfonodi e problemi di adattamento al caldo o al freddo, oltre che all’intenso esercizio fisico.
I sintomi peggiorano con l’età, principalmente a causa del progressivo accumulo di glicolipidi.
Segni e sintomi della malattia di Fabry compaiono in genere più tardi nella vita e sono più lievi nelle femmine (in una piccola percentuale di casi la malattia decorre addirittura in modo asintomatico, ovvero privo di disturbi).
Complicazioni
Il decorso della malattia è variabile, ma di solito si manifesta con
- aumento della produzione di urina (poliuria),
- aumento della sete (polidipsia),
- comparsa di proteine nelle urine (proteinuria)
dovuti a un progressivo danno renale che in ultimo condurrà ad insufficienza renale nella terza-quarta decade di vita.
Si osservano disturbi cardiaci (e pressione alta) e difetti valvolari, oltre che manifestazioni neurologiche.
Nel complesso, le possibili complicazioni comprendono:
- Cardiomegalia (ingrossamento del cuore)
- Aritmie (sviluppo di alterazioni del battito cardiaco)
- Perdita dell’udito.
L’accumulo progressivo di glicolipidi con conseguente rigonfiamento e proliferazione delle cellule endoteliali favorisce infine malattie cardiache e ictus, con possibili esiti fatali.
Diagnosi
La presenza della malattia viene sospettata in caso di sintomi compatibili con il quadro descritto, soprattutto in presenza di familiarità per la malattia; la diagnosi di conferma avviene mediante misurazione dell’attività alfa-galattosidasi-A e/o esami genetici.
Ove non disponibili, la biopsia dei tessuti (analisi di laboratorio di un piccolo campione di pelle/rene) può evidenziare la presenza di accumuli di caratteristici glicolipidi.
Cura
La malattia di Fabry non può purtroppo ad oggi essere curata, il trattamento è quindi mirato al sollievo dai sintomi e alla prevenzione delle possibili complicazioni; è possibile somministrare l’enzima carente (alfa-galattosidasi A) nei pazienti caratterizzati da un quadro grave fin dal momento della diagnosi, mediante infusioni sostitutive ogni due settimane.
Esiste anche una terapia in forma orale, Galafold (migalastat, Amicus Therapeutics), che può legarsi, stabilizzare e migliorare l’attività enzimatica residua di alcune forme.
Medicinali come gli ACE-inibitori sono i farmaci di prima scelta nei pazienti che presentino pressione alta.
L’ipertensione in questi pazienti deve essere gestita con un inibitore dell’enzima di conversione dell’angiotensina o un bloccante del recettore dell’angiotensina.[12][13][1]
I pazienti con malattia di Fabry con malattia renale allo stadio terminale devono ricorrere a dialisi e sono candidabili al trapianto.
Fonti e bibliografia
- Fabry Disease – Syed Rizwan A. Bokhari; Hassam Zulfiqar; Anis Hariz
- rarediseases.org
- MedLinePlus
- NIH
Tutti gli aggiornamenti su salute, alimentazione e benessere.






