Introduzione
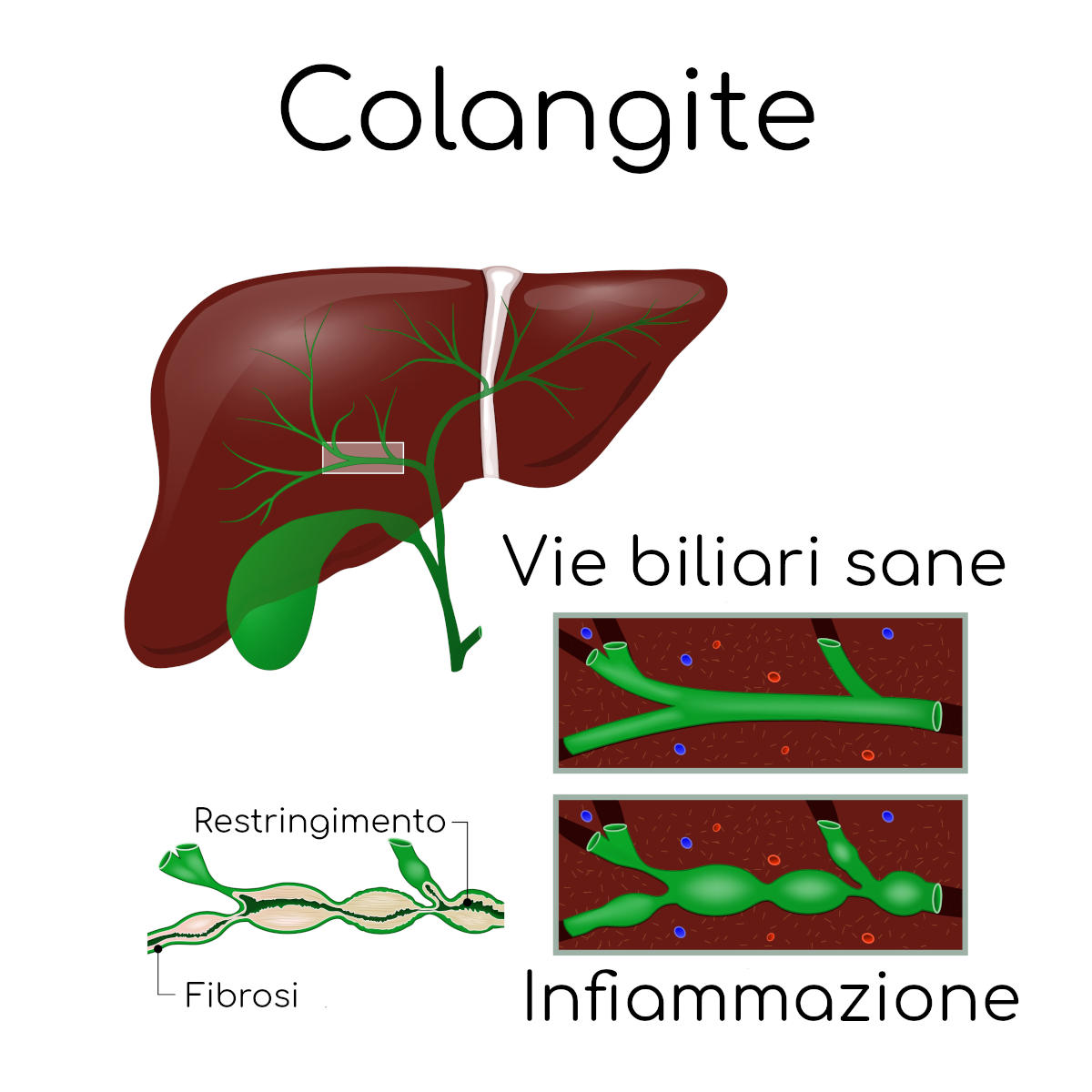
La bile è una sostanza che viene prodotta all’interno del fegato e poi inviata verso l’intestino, dove contribuisce alla digestione dei grassi introdotti con il cibo, mediante specifici vasi (dotti biliari). Se questi canali per il trasporto diventano troppo larghi la bile inevitabilmente inizierà a raccogliersi, stagnando ed accumulandosi, favorendo così lo sviluppo di infezioni (colangite).
La malattia di Caroli è anche nota come dilatazione biliare congenita intraepatica:
- dilatazione: aumento della larghezza
- biliare: riferita alle vie biliari, i canali che collegano fegato, cistifellea e intestino per il trasporto della bile
- congenita: presente sin dalla nascita
- intraepatica: riferita alla porzione relativa al fegato.
È una rara malattia genetica; nella forma classica è caratterizzata da episodi ricorrenti di infiammazione delle vie biliari (colangite) e dall’assenza di fibrosi (fenomeno in cui il normale tessuto viene sostituito da cicatrici, prive della normale e necessaria funzionalità); di norma viene diagnosticata durante l’adolescenza.
Shutterstock/Designua
È importante a questo proposito distinguere due diversi casi:
- la malattia di Caroli comporta la sola presenza delle malformazioni delle vie biliari e si verifica sporadicamente, ovvero in assenza di una storia familiare della condizione (con poche eccezioni di trasmissione autosomica dominante),
- la sindrome di Caroli si riferisce invece alla contemporanea presenza di fibrosi epatica congenita (questa condizione viene invece trasmessa geneticamente da genitori ai figli ed è spesso associata al rene policistico autosomico recessivo) e si tratta della forma più comune.
La malattia di Caroli (1 individuo su 1.000.000) è più rara della sindrome di Caroli (1 individuo su 100.000), ma si tratta di stime difficile calcolare con precisione.
Il trattamento è esclusivamente di supporto mediante antibiotici che possano curare l’infezione e, quando necessario, un trattamento endoscopico con colangiopancreatografia retrograda endoscopica per (ERCP) per risolvere un’eventuale ostruzione.
Cause
La sindrome di Caroli è una condizione estremamente rara e comunque più diffusa nelle popolazioni asiatiche.
Si tratta di una malattia genetica, ovvero la cui causa è un errore presente nel DNA (gene PKHD1), in particolare nella porzione relativa alle istruzioni per la produzione di una proteina (fibrocistina) che nel fegato riveste un importante ruolo nella gestione della composizione della bile: è in grado di rilevare stimoli meccanici, chimici e osmotici per poi modificarla di conseguenza. È anche coinvolta nel normale sviluppo del fegato (e dei reni) attraverso il controllo della proliferazione cellulare.
Le conseguenze a livello del fegato consistono nello sviluppo di gravi danni strutturali al tessuto dell’organo, che si manifestano in forma di dilatazioni anomale che alla fine portano alla fibrosi epatica, un processo di guarigione delle lesioni che tuttavia risulta fuori controllo e responsabile quindi della deposizione di un eccesso di tessuto cicatriziale al posto di quello normale.
Questa dilatazione dei vasi porta al rallentamento ed al blocco della bile nel fegato, con la formazione di calcoli biliari e successiva ostruzione, un terreno molto fertile per la comparsa di gravi infezioni (colangite).
La stessa proteina è coinvolta anche nella genesi del rene policistico autosomico recessivo, motivo per il quale le due condizioni sono spesso presenti nello stesso paziente. Poiché questa condizione presenta caratteristiche molto simili alla malattia di Caroli, è possibile che anche in quest’ultima sussista un’alterazione di questa proteina, ma ad oggi non se ne ancora la certezza.
La malattia di Caroli fu descritta per la prima volta sul finire degli anni ’50 del secolo scorso dal dottor Jacques Caroli, un gastroenterologo francese.
Trasmissione genetica della sindrome di Caroli
La sindrome di Caroli è la conseguenza di difetti genetici del gene PKHD1; quando collegata al rene policistico si osserva una trasmissione autosomica recessiva (vale la pena notare che la forma di rene policistico in questione rende conto di una minoranza di casi, mentre la più comune ha trasmissione dominante).
Le malattie trasmesse con modalità recessiva si manifestano solo quando si eredita il difetto genetico da entrambi i genitori, mentre se un individuo riceve un gene funzionante e uno difettoso questi sarà portatore sano, ma di norma senza alcun disturbo (il gene funzionante è successivo a compensare).
- Il rischio per due genitori portatori sani di trasmettere entrambi il gene non funzionante e concepire un figlio affetto è del 25% a ogni gravidanza.
- Il rischio di concepire un figlio portatore, come i genitori, è del 50% ad ogni gravidanza.
- La possibilità di ricevere due copie entrambe funzionanti è infine del 25%.
Sintomi
I sintomi di entrambe le forme esordiscono in genere all’età di 30 anni, ma possono di fatto manifestarsi a qualsiasi età.
La malattia di Caroli può colpire sia il fegato che i reni (sindrome di Caroli); pertanto, le manifestazioni cliniche della malattia appaiono tanto come complicanze renali che epatiche. Poiché i reni sono l’organo più colpito, la forma più comunemente osservata per prima è la presenza di reni policistici nel neonato (un rene policistico presenta numerose cisti, ovvero sacche piene di liquido, nel normale tessuto renale).
Le anomalie riguardanti il fegato si osservano invece più tardi nella vita, a causa di tempistiche di espressione differenti relative alla proteina coinvolta.
Poiché la malattia di Caroli è caratterizzata da un flusso della bile reso complicato dalla presenza di dilatazioni anomale, si osserva prima la formazione di fango biliare (la bile si fa più densa) fino ad una vera e propria stasi (blocco) e ostruzione. Questo predispone allo sviluppo di infezioni ricorrenti (colangite batterica) che si manifestano con
- dolore addominale,
- febbre e brividi,
- fegato ingrossato,
- ittero (colorazione gialla di pelle e parte bianca degli occhi, a causa della colestasi),
- prurito (causato da un aumento della quantità di bilirubina in circolo).
Complicazioni
Più raramente la malattia potrebbe esordire in età avanzata, dopo lo sviluppo della fibrosi epatica, con gravi sintomi e segni di ipertensione portale, come
Il fegato dispone di una propria fitta rete di vasi sanguigni che costituiscono il sistema portale epatico, responsabile del trasporto diretto di sangue dagli organi digestivi al fegato, affinché questi possa filtrare eventuali sostanze di rifiuto introdotte con il cibo. In caso di fibrosi epatica anche questo sistema va incontro a complicazioni ed alterazioni nel suo funzionamento, con un progressivo aumento della pressione del sangue al suo interno (ipertensione portale).
I pazienti con malattia di Caroli sono esposti ad aumentato rischio di sviluppare colangiocarcinoma.
Diagnosi
La malattia di Caroli viene solitamente diagnosticata dopo il primo episodio di colangite, riconosciuto attraverso i sintomi descritti.
Gli accertamenti di laboratorio mostreranno leucocitosi, ovvero un aumento dei globuli bianchi nel sangue, segno della risposta del sistema immunitario all’infezione; si rileva tipicamente anche un aumento di
Dal punto di vista degli esami di imaging l’esame di prima scelta è l’ecografia, per l’ottimo rapporto costo/qualità delle osservazioni, ma questa può essere seguita da approfondimenti come la tomografia computerizzata o esami specialistici come la colangiopancreatografia retrograda endoscopica o mediante risonanza magnetica(il vantaggio della prima è la possibilità di intervenire nella stessa seduta per la rimozione di un calcolo)
La biopsia epatica, esame più invasivo che richiede il prelievo di un piccolo campione dell’organo, viene eseguita raramente e tipicamente allo scopo di trovare conferma della presenza di fibrosi epatica associata.
Cura
La prognosi per la malattia di Caroli varia a seconda della gravità con cui si manifesta (a sua volta espressione del tipo di anomalia genetica) oltre che del numero di apparati coinvolti.
Il cardine della terapia è rappresentato dalla gestione e supporto del paziente e dei sintomi presentati; la colangite da ostruzione biliare (infezione delle vie biliari) viene trattata con antibiotici, ma alcuni pazienti richiedono un drenaggio del contenuto presente (praticato mediante tecniche mininvasive).
Dal punto di vista farmacologico l’acido ursodesossicolico può essere valutato per contrastare la formazione di bile troppo densa, mentre alcuni autori suggeriscono trattamenti chirurgici per la forme di gravi e ricorrenti della malattia di Caroli (prevedendo ad esempio una rimozione di parte del fegato, soprattutto quando la parte interessata sia concentrata su un lato).
In presenza di fibrosi epatica (sindrome di Caroli) questa purtroppo porta con sé diverse possibili complicazioni, che richiedono a loro volta una gestione mirata e puntuale (come quando conseguenti a cirrosi epatica), ad esempio mediante
- beta-bloccanti (farmaci per abbassare la pressione)
- diuretici per l’ascite,
- legatura delle varici esofagee.
Il trapianto di fegato è ad oggi l’unico trattamento definitivo disponibile per la sindrome di Caroli, che si rende necessario in caso di:
- grave scompenso epatico,
- infezione ricorrente che non risponde più alla terapia,
- sviluppo di tumore.
Fonti e bibliografia
- Caroli Disease – Jalaluddin Umar; Pujitha Kudaravalli; Savio John
- RareDiseases
Tutti gli aggiornamenti su salute, alimentazione e benessere.






