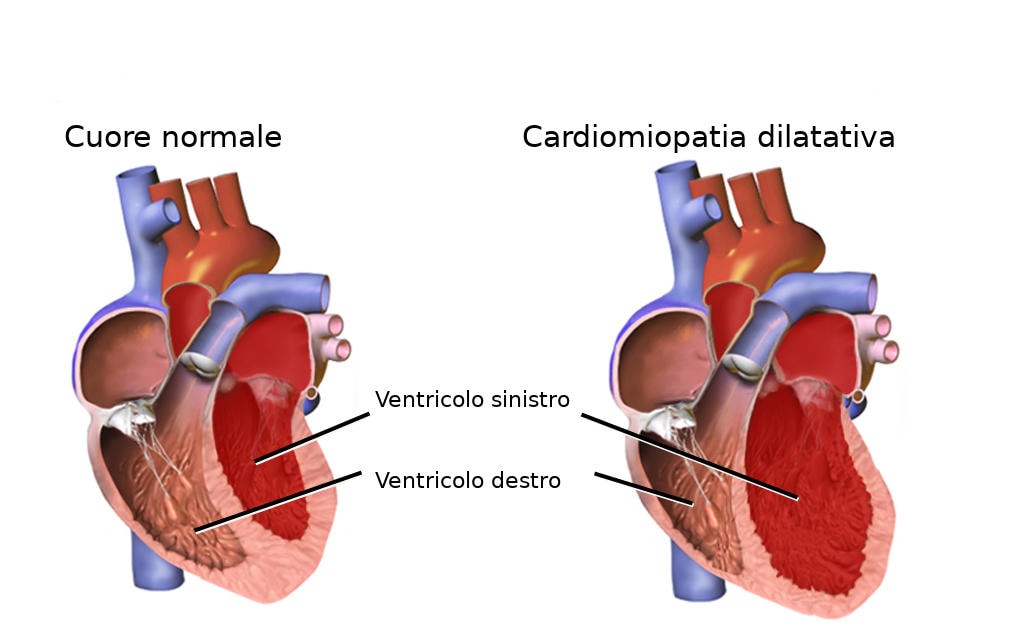
Cos’è l’amiloidosi (in parole semplici)?
L’amiloidosi è una malattia rara che si verifica quando alcune proteine, che normalmente circolano nel sangue, iniziano a comportarsi in modo anomalo: invece di mantenere la loro forma corretta, si ripiegano male e si aggregano tra loro, formando dei depositi fibrosi (chiamati amiloidi) che si accumulano in vari organi.
È come se questi depositi “impregnassero” i tessuti, impedendo agli organi di funzionare correttamente.
Ne esistono diverse forme (oltre 40) e purtroppo ad oggi non esiste una cura definitiva.
Amiloidosi da transtiretina
Una forma particolare di amiloidosi è quella causata dalla proteina transtiretina (chiamata anche TTR) nella sua forma naturale, detta “wild type”.
Questa forma, purtroppo nota per aver causato la morte del fotografo Oliviero Toscani nel gennaio del 2025, colpisce principalmente le persone anziane, soprattutto uomini sopra i 65 anni.
La transtiretina è una proteina prodotta dal fegato che normalmente trasporta ormoni tiroidei e vitamina A nel sangue.
Con l’avanzare dell’età, nei pazienti colpiti dalla malattia, questa proteina diventa instabile: è come se i “mattoncini” che la compongono si staccassero e si riattaccassero in modo disordinato, formando delle fibrille che si depositano principalmente nel cuore.
I pazienti affetti da amiloidosi cardiaca possono sviluppare tra l’altro disturbi come
- un battito cardiaco anomalo (aritmia),
- un cuore ingrossato (cardiomegalia)
- e ipertensione ortostatica.
Questo accumulo progressivo rende il muscolo cardiaco più rigido e meno efficiente nel suo lavoro di pompa (insufficienza cardiaca). Poiché questa forma di amiloidosi è legata all’invecchiamento naturale della proteina (e non a mutazioni genetiche), viene chiamata “wild type” o “senile”.
Cause
Esistono diverse forme di amiloidosi, che dipendono dal tipo di proteina depositata e da quali tessuti sono coinvolti; nel 90% dei casi i depositi di fibrille sono formati da proteina amiloide dalla struttura alterata che si trasforma in aggregati insolubili per poi accumularsi, mentre nel 10% sono costituiti da altre molecole (ad esempio cheratina, calcitonina e fattore natriuretico atriale).
Si pensa che le forme localizzate della malattia siano causate da una produzione locale delle proteine responsabili della precipitazione, più che da una deposizione di quelle presenti nel sangue; molti meccanismi contribuiscono alla precipitazione degli aggregati proteici nei tessuti e quindi alla genesi della malattia, come:
- patologica degradazione delle proteine (proteolisi),
- proteolisi deficitaria o assente,
- mutazioni genetiche che provocano alterazioni delle proprietà termodinamiche e di trasporto delle proteine,
- infezioni croniche,
- altri meccanismi non ancora pienamente definiti.
Classificazione
Esistono circa trenta diversi tipi di amiloidosi, in base alla forma, la funzione, la sequenza di amminoacidi e la struttura delle fibrille coinvolte.
Le più frequenti amiloidosi sono:
- Amiloidosi da catene leggere (AL – amyloid light chains). Anche chiamata amiloidosi primaria, è il tipo più comune ed è causata dall’accumulo di immunoglobuline (anticorpi) formate da catene leggere. Anche se la causa esatta non è nota, questo tipo insorge quando il midollo osseo produce un numero elevato di anticorpi che non possono essere smaltiti. Questa condizione è associata al mieloma multiplo, un tipo di tumore che colpisce le cellule del sangue. Le catene leggere si depositano nei reni, cuore, fegato, intestino e neuroni.
- Amiloidosi reattiva a infiammazione cronica (AA – amyloid type A protein). In passato denominata amiloidosi secondaria, è causata da alcune condizioni infiammatorie croniche come l’artrite reumatoide, il morbo di Crohn o la rettocolite ulcerosa. Colpisce principalmente i reni, ma può interessare anche l’apparato digerente, il fegato e il cuore.
- Amiloidosi correlata a dialisi (DRA – dialysis related amyloidosis). A causa della dialisi è il tipo più comune nelle persone anziane o che sono state in dialisi per più di 5 anni. La proteina coinvolta è la beta2-microglobulina e si deposita più frequentemente a livello delle ossa, articolazioni e tendini.
- Amiloidosi familiari o ereditarie (ATTR, AAPOAI, ecc). Queste forme sono condizioni rare trasmesse per via ereditaria che spesso coinvolgono fegato, neuroni, cuore e reni. Esistono diverse mutazioni genetiche che possono predisporre allo sviluppo della malattia, ad esempio la mutazione della proteina transtiretina (TTR) o dell’apolipoproteina AI (APOAI).
- Amiloidosi senile. È determinata dall’accumulo della proteina TTR non mutata (wild-type) a livello cardiaco, dei nervi e in altri tessuti. È più frequente nel sesso maschile e legata all’invecchiamento.
- Amiloidosi organo-specifica. Si tratta di una forma che si verifica quando le proteine si accumulano nei singoli organi, come ad esempio l’amiloidosi cutanea da accumulo di cheratina, il carcinoma midollare della tiroide dovuto ad accumulo di calcitonina, o l’amiloidosi atriale dovuta all’accumulo di fattore natriuretico atriale a livello cardiaco.
Fattori di rischio
I fattori di rischio per lo sviluppo della malattia sono rappresentati da:
- sesso maschile (molto comune nelle forme AL e ATTR wild-type),
- età avanzata (soprattutto sopra i 65-70 anni),
- presenza di malattie concomitanti come le infezioni croniche (per l’amiloidosi AA) o il mieloma multiplo (per l’amiloidosi AL),
- presenza di insufficienza renale cronica e dialisi (amiloidosi correlata a dialisi DRA).
Sintomi

Shutterstock/PeopleImages.com – Yuri A
I sintomi dell’amiloidosi sono estremamente variabili in base ai tessuti colpiti, ma le manifestazioni sistemiche più comuni sono relative al coinvolgimento di cuore e rene:
- aritmie (irregolarità del battito cardiaco),
- dispnea (difficoltà di respirazione),
- fatica,
- gonfiore delle caviglie (edema),
- anemia,
- ritenzione idrica,
- perdita di proteine nelle urine (proteinuria).
Tra gli altri possibili sintomi ricordiamo invece:
- variazione del colore della pelle,
- decadimento cognitivo,
- sensazione di pienezza,
- dolore articolare,
- rigonfiamento della lingua,
- difficoltà nella deglutizione,
- formicolio e intorpidimento di gambe e piedi,
- debolezza nelle mani,
- perdita di peso improvvisa.
Amiloidosi cardiaca
Il deposito di fibrille a livello cardiaco può causare un irrigidimento delle pareti cardiache e una diminuzione della funzionalità e dell’attività elettrica del cuore.
In situazioni gravi potrebbe verificarsi insufficienza cardiaca perché i muscoli della parete non sono più in grado di pompare il sangue in maniera adeguata.
I sintomi caratteristici sono
- aritmie;
- segni di scompenso cardiaco:
- caviglie e piedi gonfi (edema),
- debolezza,
- fatica,
- dispnea,
- nausea,
- …
Amiloidosi renale
Quando le proteine si depositano a livello del rene, possono comprometterne la funzione di filtro. Il rene, infatti, elimina le sostanze tossiche e l’acqua in eccesso e, se questa funzione è interrotta, si accumulano nell’organismo provocando:
- segni di insufficienza renale:
- aumento della creatinina nel sangue,
- ritenzione idrica che si manifesta con
- una diminuzione della produzione di urina,
- edemi alle caviglie, ai piedi e intorno agli occhi;
- perdita nelle urine delle proteine importanti per il corpo (proteinuria);
Amiloidosi gastro-intestinale
I depositi di amiloide a livello intestinale interferiscono con le funzioni di trasporto, digestione e assimilazione del cibo portando a:
- perdita dell’appetito,
- diarrea,
- nausea,
- gastralgia (dolore allo stomaco),
- perdita di peso,
- epatomegalia (ingrossamento del fegato).
Neuropatia amiloide (amiloidosi cerebrale)
Quando l’amiloidosi colpisce il cervello, i nervi periferici e il midollo spinale, le informazioni raccolte dall’ambiente esterno non sono integrate a livello cerebrale. Si manifesteranno quindi problemi di
- equilibrio,
- controllo della vescica e dell’intestino,
- sudorazione,
- formicolio e stanchezza a gambe e braccia,
- ipotensione (pressione bassa) e svenimenti,
- decadimento cognitivo.
Complicazioni
Negli ultimi anni sono stati fatti straordinari progressi nella terapia delle amiloidosi e la prognosi dei pazienti è in continuo miglioramento.
Possono presentarsi comunque alcune complicazioni anche gravi:
- Renali:
- proteinuria (perdita di proteine nelle urine),
- ritenzione idrica e di sostanze tossiche fino all’insufficienza renale;
- Cardiache:
- aritmie (anomalie nella conduzione elettrica del cuore),
- ipertrofia delle pareti con irrigidimento e diminuzione della funzione di pompa fino allo scompenso cardiaco;
- Neuronali:
- dolore,
- intorpidimento e formicolio,
- mancanza di sensibilità e bruciore ai piedi o alle dita,
- alterazioni dell’alvo (alternanza di stitichezza e diarrea) se è coinvolta l’innervazione intestinale,
- ipotensione e svenimento se è colpita l’innervazione che controlla la pressione arteriosa,
- decadimento cognitivo.
Diagnosi
Identificare l’amiloidosi rappresenta spesso una sfida clinica complessa, poiché i sintomi iniziali sono comuni a molte altre patologie. Tuttavia, i protocolli diagnostici attuali permettono oggi una precisione senza precedenti, spesso riducendo la necessità di procedure invasive.
Il sospetto clinico sorge solitamente in presenza di un’insufficienza cardiaca inspiegabile, di una proteinuria persistente o di una neuropatia periferica. L’iter diagnostico moderno segue due percorsi principali a seconda della proteina sospettata:
Screening iniziale e biomarcatori
Il primo passo consiste nell’escludere o confermare la presenza di una componente monoclonale (proteine anomale prodotte dal midollo osseo). Questo viene effettuato tramite:
- Immunofissazione sierica ed urinaria: per rilevare frammenti di anticorpi.
- Dosaggio delle catene leggere libere (FLC): un test del sangue fondamentale per la diagnosi di amiloidosi AL.
- Biomarcatori cardiaci: il dosaggio di NT-proBNP e troponina non serve solo alla diagnosi, ma è cruciale per definire lo stadio di gravità e la prognosi.
Imaging avanzato e tecniche non invasive
Negli ultimi anni, la diagnosi di amiloidosi cardiaca da transtiretina (ATTR) ha subito una rivoluzione grazie a tecniche di imaging che permettono di evitare la biopsia cardiaca in molti pazienti:
- Scintigrafia ossea con traccianti difosfonati: questo esame è diventato il cardine diagnostico per la forma ATTR. Se il cuore “capta” intensamente il tracciante e i test per la componente monoclonale sono negativi, la diagnosi di amiloidosi ATTR è confermata con un’accuratezza vicina al 100%.
- Risonanza magnetica cardiaca (RMC): attraverso il “Late Gadolinium Enhancement” (LGE) e la mappatura T1, la risonanza magnetica è in grado di visualizzare i depositi di amiloide tra le cellule del cuore, mostrando un pattern tipico molto specifico.
- Ecocardiogramma con studio dello strain: una tecnica ecografica avanzata che mostra come il cuore si contrae; nell’amiloidosi è tipico il risparmio dell’apice del cuore rispetto alle altre zone (pattern “cherry on top”).
Conferma istologica e analisi genetica
La biopsia rimane il “gold standard” quando i test non invasivi non sono conclusivi o quando si sospetta una forma AL. Il campione viene trattato con il colorante Rosso Congo: se osservato al microscopio a luce polarizzata, l’amiloide mostra una tipica birifrangenza verde mela.
Una volta confermata la presenza di amiloide, è essenziale la tipizzazione (identificare quale proteina sta causando il problema) tramite immunoflorescenza o spettrometria di massa. Infine, in caso di amiloidosi ATTR, viene sempre eseguita l’analisi genetica per distinguere la forma “wild-type” (senile) dalla forma ereditaria, informazione fondamentale per i familiari del paziente.
Cura
Il trattamento dell’amiloidosi ha l’obiettivo primario di arrestare la produzione della proteina tossica e proteggere la funzione degli organi colpiti. Grazie alle recenti innovazioni, disponiamo oggi di terapie mirate che hanno radicalmente cambiato le prospettive di vita dei pazienti.
Trattamento dell’amiloidosi AL (da catene leggere)
In questa forma, la cura è diretta contro le plasmacellule del midollo osseo che producono le catene leggere anomale. L’approccio attuale prevede:
- Immunoterapia e chemioterapia: il protocollo di prima linea standard combina farmaci biologici come il daratumumab (un anticorpo monoclonale anti-CD38) con regimi chemioterapici (bortezomib, ciclofosfamide e desametasone). Questa combinazione permette di ottenere risposte rapide e profonde.
- Trapianto di cellule staminali: per i pazienti giovani e con una funzione cardiaca ancora preservata, il trapianto autologo di cellule staminali rimane un’opzione per consolidare i risultati della chemioterapia.
Trattamento dell’amiloidosi ATTR (da transtiretina)
Per la forma da transtiretina, sia ereditaria che wild-type, la strategia si basa su tre fronti:
- Stabilizzatori della proteina: farmaci come il tafamidis si legano alla transtiretina nel sangue, impedendole di sfaldarsi e formare depositi. È attualmente la terapia d’elezione per l’amiloidosi cardiaca ATTR, dimostratasi efficace nel ridurre la mortalità e i ricoveri.
- Silenziatori genici (RNA-interference e Antisense): farmaci innovativi come vutrisrisan, patisiran ed eplontersen agiscono “spegnendo” il messaggio genetico nel fegato. Questo blocca quasi totalmente la produzione della proteina TTR. Sono utilizzati principalmente per le forme con coinvolgimento neurologico, ma il loro impiego si sta estendendo con successo anche alla forma cardiaca.
- Terapie emergenti: sono in fase avanzata di studio anticorpi capaci di “aggredire” e rimuovere i depositi di amiloide già presenti nei tessuti, oltre a tecnologie di editing genetico (CRISPR) per una correzione definitiva del difetto alla fonte.
Terapie di supporto e stile di vita
La gestione dei sintomi è altrettanto importante quanto la terapia specifica. Poiché il cuore amiloide è rigido, la gestione dei liquidi è critica: i pazienti devono spesso seguire una dieta iposodica (poco sale) e monitorare quotidianamente il peso corporeo per regolare l’uso dei diuretici. È fondamentale prestare attenzione ai farmaci comuni per la pressione (come beta-bloccanti o ACE-inibitori), che a volte possono essere mal tollerati in questi pazienti.
Un’attività fisica leggera e costante, compatibile con le riserve cardiache, è incoraggiata per mantenere la forza muscolare e migliorare il benessere generale. In casi estremi e selezionati, si può ricorrere al trapianto di fegato (per interrompere la produzione di proteina mutata) o al trapianto di cuore, sebbene queste opzioni siano oggi meno frequenti grazie all’efficacia dei nuovi farmaci.
Il supporto multidisciplinare che coinvolge cardiologi, ematologi, neurologi e nefrologi è essenziale per coordinare i controlli periodici (ECG, esami del sangue, ecocardiogrammi) e adattare la terapia in tempo reale, garantendo la migliore qualità di vita possibile.
Fonti e bibliografia
Tutti gli aggiornamenti su salute, alimentazione e benessere.