Cosè l’acromegalia?
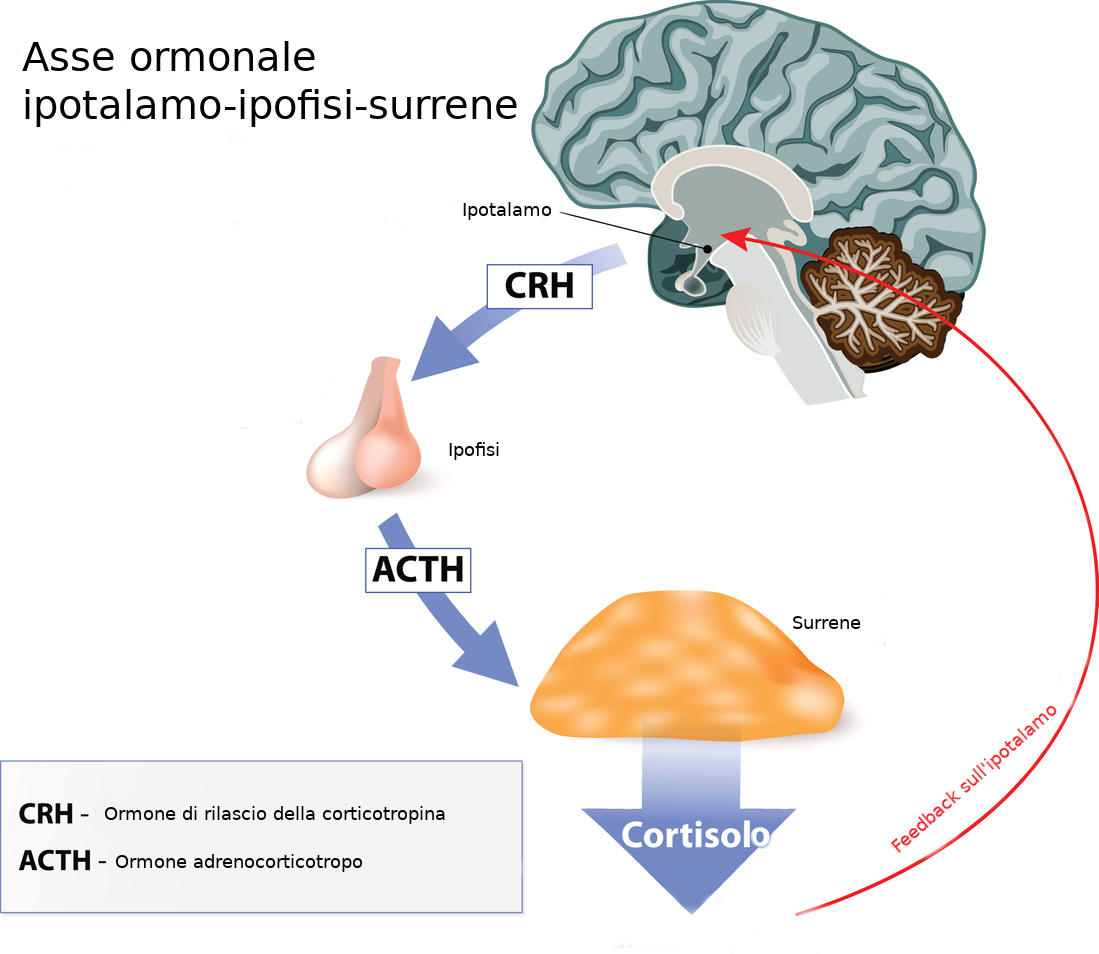
L’acromegalia (dal greco “acros” e “megalos” ,a significare “grandi estremità”) è una rara patologia endocrinologica cronica, derivante dall’esposizione dell’organismo a grandi quantità dell’ormone della crescita GH, prodotto nella stragrande maggioranza dei casi da un adenoma dell’ipofisi (tumore benigno).
A seconda dell’età in cui si verifica questa ipersecrezione di ormone della crescita si parlerà di:
- gigantismo, quando si verifica nel bambino o adolescente in fase di crescita;
- acromegalia, quando si verifica nell’adulto dove, per via dell’ormai avvenuta fusione delle cartilagini epifisarie a livello osseo, il soggetto non potrà più crescere in altezza.
L’acromegalia è una malattia relativamente rara, poiché colpisce circa 40 individui su 1 milione, con un rapporto maschi/femmine di 1 a 1. L’incidenza è di circa 4 nuovi casi all’anno ogni milione di abitanti, con una diagnosi che avviene con circa 10 anni di ritardo ad un’età media di 40-45 anni.
Le cause di acromegalia sono da ricercare nella presenza di:
- adenoma ipofisario che iper-produce GH (nel 95% dei casi),
- secrezione ectopica di GHRH da parte dell’ipotalamo o di un tumore periferico,
- secrezione ectopica di GH (molto rara) da parte di un tumore polmonare, pancreatico o di un linfoma.
Il quadro clinico dell’acromegalia è caratterizzata da diversi sintomi piuttosto caratteristici:
- ingrossamento dei tessuti molli e delle ossa con:
- prominenza delle arcate sovraorbitarie, della fronte, degli zigomi e della mandibola,
- ingrossamento del naso e della lingua,
- ispessimento della cute, ipertricosi e sudorazione eccessiva,
- comorbidità metaboliche e sistemiche con sviluppo di:
- ipertensione,
- cardiopatia congestizia (da cardiomegalia),
- diabete,
- dislipidemia e sindrome metabolica,
- riduzione della normale secrezione ipofisaria con riduzione dei livelli di TRH, ACTH, FSH e LH.

By Philippe Chanson and Sylvie Salenave – Acromegaly. Orphanet Journal of Rare Diseases 2008, 3:17. doi:10.1186/1750-1172-3-17, CC BY 2.0, Link
La diagnosi si basa inizialmente su anamnesi ed esame obiettivo coadiuvati dal controllo biochimico di GH ed IGF-1. Come secondo livello è richiesta l’esecuzione di una risonanza magnetica per individuare la causa organica dell’iper-produzione di GH, che nella quasi totalità dei casi è dovuta ad un adenoma a carico dell’ipofisi.
Il trattamento dell’acromegalia prevede la rimozione della causa sottostante, per esempio in caso di adenoma ipofisario è possibile:
- un approccio farmacologico con gli analoghi della somatostatina che riducono la secrezione e gli effetti del GH,
- radioterapia,
- intervento chirurgico di asportazione del tumore ipofisario, con conservazione della funzionalità ipofisaria residua.
Oltre al trattamento dell’adenoma risulta fondamentale la gestione e il trattamento delle comorbidità come ipertensione, scompenso cardiaco, insufficienza respiratoria, diabete, dislipidemia con aterosclerosi.
Poiché la condizione è rara e i sintomi compaiono in modo molto graduale, talvolta la condizione richiede molto tempo prima di destare sospetti ed essere riconosciuta; un eccessivo ritardo può tuttavia avere complicazioni notevoli. la prognosi dell’acromegalia dipende infatti dalle possibilità del trattamento, che deve essere impostato precocemente subito dopo aver definir la diagnosi con certezza. In caso di trattamento tempestivo ed efficace è possibile osservare una netta riduzione della mortalità dovuta alla malattia.
Cause
Nel soggetto sano il GH (ormone della crescita) viene rilasciato con picchi secretori che si verificano durante la notte (specie durante la fase REM del sonno), tipicamente nel bambino e nel soggetto in età evolutiva, per consentire la crescita staturale. La secrezione di GH è quindi pulsatile (intermittente) e, al di là di tali picchi, la produzione basale di GH è molto bassa; questi picchi tendono poi a diventare sempre più rari nella vita adulta sino ad avere livelli di GH indosabili.
In caso di acromegalia non si osservano alti livelli di GH, ma ciò che accade è che si verifica una secrezione continua e costante, con assenza di regolazione, poiché essa viene a svincolarsi dai meccanismi neuro-ormonali previsti. La conseguenza è che la quantità di GH in circolo è stabilmente sopra la soglia di 1 ng/mL, valore che permette di discriminare una situazione normale da quella patologica.
La principale causa di tale secrezione non pulsatile persistentemente aumentata è dovuta nel 98% dei casi ad un adenoma ipofisiario, un tumore benigno che potrà essere formato dalle sole cellule somatotrope oppure essere misto (con iperprolattinemia consensuale, ossia una contemporanea eccessiva produzione di prolattina).
L’aumentata produzione di GH stimola la produzione del suo ormone messaggero, attraverso il quale esercita gran parte dei suoi effetti biologici: l’ormone in questione è il IGF-1 (Insuline-like Growth Factor-I), anche detto somatomedina, prodotto a livello epatico.
Dal punto di vista eziologico si distinguono:
- Forme primitive (98% dei casi):
- da adenomi ipofisari a cellule somatotrope; di solito si tratta di macroadenomi che superano 1 cm di diametro;
- adenomi ipofisari misti con secrezione associata di GH e prolattina.
- Forme secondarie (2% dei casi):
- da secrezione ectopica di GHRH da carcinoidi o tumore delle isole di Langherans del pancreas;
- da secrezione ectopica di GHRH (ormone di stimolazione del GH prodotto normalmente dall’ipotalamo) da parte di tumori dell’ipotalamo;
- da secrezione ectopica di GH da parte di tumori polmonari, pancreatici o di un linfoma.
Sintomi
Dal punto di vista clinico si descrive una sintomatologia suddivisibile in 3 gruppi principali:
- Sintomi dovuti all’espansione del tumore a livello dell’ipofisi, che può portare allo sviluppo di:
- cefalea (mal di testa),
- vomito di tipo centrale a getto e senza nausea,
- ipertensione endocranica,
- disturbi visivi (come l’emianopsia bitemporale, che causa la perdita di metà del campo visivo);
inoltre l’adenoma in crescita tende a danneggiare le cellule ipofisarie contigue che erano normofunzionanti, portando ulteriori alterazioni ormonali (si osserva in particolare un quadro di ipo-pituitarismo con riduzione della secrezione di TSH, FSH e LH (gonadotropine) o ACTH con i relativi sintomi associati.
- Sintomi dovuti all’effetto diretto del GH in eccesso, che provoca insulino-resistenza e intolleranza ai carboidrati con forma iniziale di diabete secondario.
- Sintomi dovuti all’IGF-1, responsabile della stimolazione a livello del DNA della proliferazione del tessuto osseo, della cartilagine, dei tessuti molli e degli altri organi portando alle tipiche manifestazioni dell’acromegalia.
Sintomi classici dell’acromegalia
Descriviamo in primis i tipici sintomi “morfologici”:
- Aumento e ipertrofia dei tessuti molli che interessa prevalentemente il volto e gli arti.
- Alterazione delle estremità ovvero mani e piedi: tipica è la deformazione “a spatola” delle dita”; molto caratteristica di questo disturbo è anche la necessità dei soggetti affetti di cambiare misura di guanti e scarpe o di allargare anelli e bracciali.
- Aumento dei seni paranasali frontali con prominenza delle arcate sovra-orbitarie
- Ingrossamento del naso e della lingua (macroglossia), che modificano il timbro della voce (anche per ispessimento della laringe) e possono portare ad un apnea ostruttiva durante il riposo.
- Ingrossamento della mandibola con prognatismo (prominenza in avanti della mandibola rispetto all’osso mascellare).
- Allargamento degli spazi interdentali.
- Ispessimento della cute (soprattutto del cuoio capelluto) che diventa seborroica, con comparsa di acne e cisti sebacee.
- Acanthosis nigricans (colorazione nerastra) a livello di ascelle e collo.
- Ipertricosi (aumento della quantità di peli), soprattutto nelle donne dove è più facilmente. riscontrabile la differenza rispetto ad un quadro di peluria fisiologica.
- Intolleranza al caldo e iperidrosi (sudorazione eccessiva) per ipertrofia delle ghiandole sudoripare.
- Dolori articolari (artralgie) e sindrome del tunnel carpale con compressione del nervo mediano a livello dei polsi.
Oltre a questi sintomi dovuti ad ingrossamento dei tessuti molli e delle ossa, si riscontrano anche “sintomi sistemici”:
- Astenia, malessere generalizzato con sonnolenza ed aumento ponderale.
- Ipertensione (pressione alta) per la ritenzione di sodio a livello renale.
- Intolleranza al glucosio per gli effetti insulino-antagonisti del GH.
- Iperinsulinemia per l’insulina-resistenza provocata sempre dal GH.
- Quadro di diabete conclamato (più raro).
- Ipogonadismo (testicoli od ovaie non producono più ormoni a sufficienza.), secondario alla compressione ipofisaria da parte dell’adenoma.
- Galattorrea (secrezione anomala di latte) da iperprolattinemia nelle donne, ginecomastia negli uomini (aumento delle dimensioni delle ghiandole mammarie).
- RIduzione del desiderio sessuale, alterazioni del ciclo mestruale nelle donne e disfunzione erettile nell’uomo.
- Organomegalia generale: ingrossamento di vari organi quali tiroide (con sviluppo di gozzo), cuore (con cardiomegalia e rischio di scompenso), fegato e milza, ghiandole salivari (con scialorrea), prostata.
Complicazioni
Col passare degli anni l’alterato quadro clinico-ormonale porta allo sviluppo di complicanze notevoli che prevedono la possibile comparsa di:
- artrite degenerativa e disabilitante, con difficoltà alla deambulazione,
- aumento del rischio di mortalità dovuto all’aterosclerosi e alla cardiopatia, all’ipertensione e al diabete,
- aumento dell’incidenza delle neoplasie (rischio elevato soprattutto per lo sviluppo di tumore al colon).
Diagnosi
La diagnosi di acromegalia è spesso tardiva a causa della progressione lenta e insidiosa dei sintomi. Il sospetto clinico nasce dall’osservazione dei cambiamenti somatici (lineamenti del volto, dimensioni di mani e piedi), ma la conferma richiede un percorso biochimico e strumentale rigoroso secondo i protocolli internazionali più recenti.
Test biochimici di primo livello
Il primo passo è il dosaggio dei livelli sierici di IGF-1 (fattore di crescita insulino-simile 1). A differenza del GH, i cui livelli fluttuano drasticamente durante il giorno, l’IGF-1 rimane stabile ed è il miglior indicatore dell’attività complessiva della malattia. Un valore di IGF-1 normale per l’età e il sesso del paziente esclude quasi sempre l’acromegalia.
Test di soppressione del GH dopo carico orale di glucosio (OGTT)
Se i livelli di IGF-1 sono elevati, si procede con il test di soppressione mediante carico orale di glucosio, considerato il “gold standard” diagnostico. Al paziente viene somministrata una soluzione contenente 75 grammi di zucchero e si misurano i livelli di GH a intervalli regolari (solitamente ogni 30 minuti per 2 ore). In una persona sana, l’iperglicemia sopprime la produzione di GH al di sotto di 0,4 µg/L (o 1,0 µg/L a seconda della sensibilità dei test locali). Nei pazienti acromegalici, questa soppressione non avviene o è insufficiente.
Localizzazione e imaging
Una volta confermata la diagnosi biochimica, è indispensabile eseguire una risonanza magnetica (RMN) della regione ipotalamo-ipofisaria con mezzo di contrasto. Questo esame permette di:
- Identificare la presenza di un adenoma (nella maggior parte dei casi un macroadenoma superiore a 10 mm).
- Valutare i rapporti del tumore con le strutture circostanti, come i nervi ottici e i seni cavernosi, informazione cruciale per la pianificazione chirurgica.
In rari casi in cui la RMN ipofisaria risulti negativa nonostante i test biochimici positivi, si ricercano fonti ectopiche di produzione di GH o GHRH (ormone stimolante il GH) tramite TC o PET total body, sospettando tumori carcinoidi polmonari o pancreatici.
Valutazione delle complicanze al momento della diagnosi
Data l’elevata frequenza di comorbidità, il protocollo diagnostico include oggi obbligatoriamente:
- Ecocardiogramma per valutare l’eventuale ipertrofia cardiaca.
- Polisonnografia per escludere apnee notturne.
- Colonscopia di screening, a causa del maggior rischio di polipi e carcinomi del colon.
- Monitoraggio metabolico (glicemia, profilo lipidico).
Cura
L’obiettivo primario della terapia è normalizzare i livelli di IGF-1 e ridurre il GH a valori di sicurezza, minimizzando così il rischio di mortalità e migliorando i sintomi. Il piano di cura è personalizzato in base alle dimensioni del tumore, ai livelli ormonali e alle condizioni generali del paziente.
Chirurgia ipofisaria
La chirurgia rappresenta la terapia di prima scelta per la maggior parte dei pazienti. L’intervento d’elezione è l’asportazione transfenoidale dell’adenoma, eseguita oggi preferibilmente per via endoscopica attraverso le cavità nasali. Questo approccio minimamente invasivo permette una rapida riduzione dei livelli ormonali e dei sintomi da compressione (come il mal di testa o i disturbi della vista). Se il tumore è confinato (microadenoma), la probabilità di guarigione definitiva è molto alta.
Terapia farmacologica
Se la chirurgia non è risolutiva o non è praticabile, la terapia medica è fondamentale. Le opzioni attuali includono:
- Analoghi della somatostatina (SSA): Sono i farmaci più utilizzati (Octreotide, Lanreotide, Pasireotide). Agiscono legandosi ai recettori dell’ipofisi, inibendo la produzione di GH e, in molti casi, riducendo le dimensioni del tumore.
- Antagonisti del recettore del GH (Pegvisomant): Questo farmaco blocca l’azione del GH sui tessuti periferici. È estremamente efficace nel normalizzare i livelli di IGF-1, anche quando gli altri farmaci falliscono.
- Agonisti della dopamina (Cabergolina): Possono essere utili nei casi più lievi o come terapia aggiuntiva, specialmente se l’adenoma produce anche prolattina.
Radioterapia e radiochirurgia
Viene riservata ai casi in cui né la chirurgia né i farmaci riescono a controllare la malattia. Tecniche moderne come la Gamma Knife o la CyberKnife permettono di colpire il residuo tumorale con estrema precisione, riducendo i danni ai tessuti cerebrali sani circostanti. Tuttavia, l’effetto completo sulla produzione ormonale può richiedere diversi anni per manifestarsi.
Stile di vita e gestione multidisciplinare
Il trattamento dell’acromegalia non si esaurisce con il controllo ormonale. È essenziale una gestione attiva delle conseguenze della malattia:
- Monitoraggio metabolico: È prioritario gestire il diabete e l’ipertensione, poiché le malattie cardiovascolari restano la principale causa di complicanze.
- Salute ossea e articolare: L’attività fisica regolare, preferibilmente a basso impatto (come nuoto o pilates), è consigliata per mantenere la mobilità articolare e contrastare l’artropatia acromegalica.
- Alimentazione: Una dieta bilanciata è fondamentale per il controllo del colesterolo e del peso corporeo, riducendo il carico sulle articolazioni già stressate dalla malattia.
- Supporto psicologico: I cambiamenti nell’aspetto fisico e la cronicità della condizione possono avere un impatto significativo sull’autostima e sul tono dell’umore.
Fonti e bibliografia
- Harrison – Principi Di Medicina Interna Vol. 1 (17 Ed. McGraw Hill)
- Malattie del sistema endocrino e del metabolismo – di G. Faglia, P. Beck-Peccoz, A. Spada
Tutti gli aggiornamenti su salute, alimentazione e benessere.