Introduzione
La fibrosi cistica (FC), o mucoviscidosi, è una malattia genetica complessa e multisistemica in grado di causare gravi danni ai polmoni e rilevanti carenze nutrizionali.
È una patologia ereditaria che colpisce le cellule che producono liquidi quali:
- muco,
- sudore,
- saliva,
- succhi gastrici.
Se in condizioni normali queste secrezioni sono fluide, nei pazienti affetti da fibrosi cistica un gene difettoso le fa diventare più spesse e viscose; anziché funzionare come lubrificanti le secrezioni otturano i condotti, i canali e i passaggi, soprattutto a livello di pancreas e polmoni.
La conseguenza più pericolosa della fibrosi cistica è l’insufficienza respiratoria, inoltre le secrezioni bloccano gli enzimi pancreatici che contribuiscono alla digestione dei grassi e delle proteine, impedendo all’organismo di assorbire le vitamine fondamentali e queste difficoltà emergono fin dall’infanzia.
Negli ultimi anni, l’avvento di terapie farmacologiche innovative ha radicalmente trasformato il decorso della malattia, permettendo di agire direttamente sulla causa genetica e non solo sui sintomi.
iStock.com/DERO2084
Aspettativa di vita
L’aspettativa di vita per le persone con fibrosi cistica è notevolmente migliorata negli ultimi decenni grazie ai progressi nella diagnosi precoce e nelle terapie molecolari. Se negli anni ’50 la maggior parte dei bambini non raggiungeva l’età scolare, oggi le proiezioni statistiche indicano che la maggior parte dei nuovi nati può aspirare a raggiungere e superare la sesta decade di vita, con una qualità della quotidianità impensabile fino a pochi anni fa.
Gravidanza
Se siete incinta o state cercando di avere un bambino, potreste prendere in considerazione l’idea di chiedere al vostro medico un esame per la fibrosi cistica per voi e per il vostro compagno. In passato i medici prescrivevano l’esame solo per le coppie maggiormente a rischio (quelle con precedenti famigliari o personali di fibrosi cistica), invece oggi l’esame è consigliato per tutte le coppie.
L’esame, eseguito in laboratorio su un campione di sangue o saliva, può aiutare a scoprire se voi o il vostro partner siete portatori del gene della fibrosi cistica. Se siete incinta e l’esame evidenzia che vostro figlio potrebbe essere a rischio di fibrosi cistica, il medico potrà consigliare ulteriori esami per il vostro bambino. La fibrosi cistica non può essere curata prima della nascita, quindi lo scopo di questi esami è quello di aiutarvi a decidere per il vostro futuro.
La decisione di sottoporsi a esami per la fibrosi cistica è del tutto personale e dipende da molti fattori, tra cui il livello di rischio e la vostra confessione religiosa.
Va inoltre tenuto presente che alcune mutazioni genetiche della fibrosi cistica non possono essere individuate dall’esame, così com’è concepito attualmente. In rari casi, quindi, potreste essere portatori della malattia, nonostante l’esame risulti normale (le possibilità che ciò avvenga sono molto basse, ma non nulle).
Cause
Nella fibrosi cistica un gene difettoso altera una proteina che regola il normale trasporto del sale (cloruro di sodio) attraverso la membrana cellulare: la conseguenza è la produzione di secrezioni più spesse e viscose nell’apparato respiratorio, in quello digerente e in quello riproduttivo. Questa malattia provoca anche una maggior concentrazione di sale nel sudore.
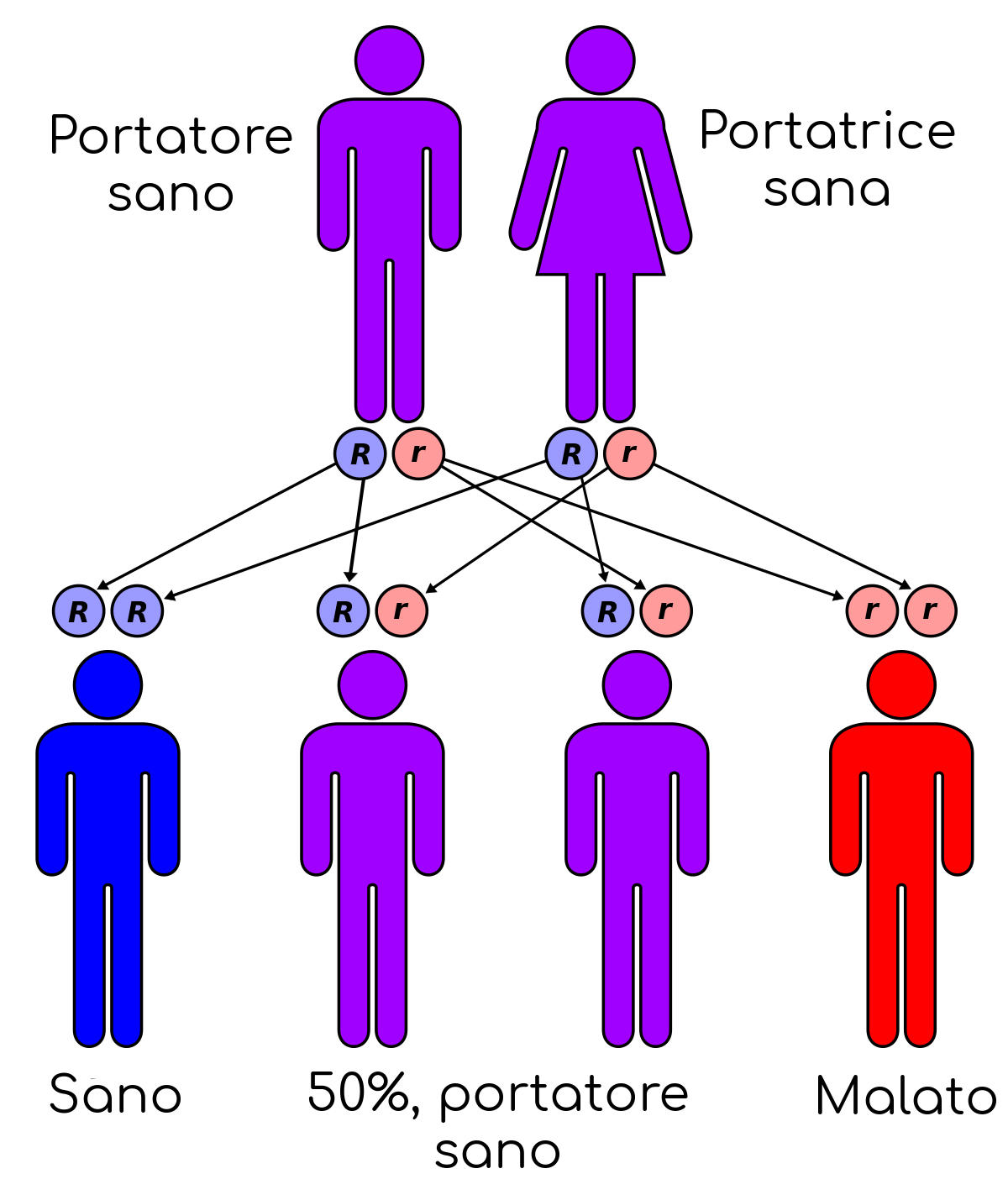
Il gene malato, ereditato dai genitori, è recessivo; questo significa che i bambini, per ammalarsi, devono ereditare due copie del gene, una per ciascun genitore. Se ereditano una sola copia, non si ammaleranno di fibrosi cistica, ma diventeranno portatori sani della malattia e la potranno trasmettere ai loro figli.
Se due persone portatrici del gene difettoso (non necessariamente malate) concepiscono un figlio, c’è il 25 per cento di probabilità che il bambino risulti ammalato di fibrosi cistica, il 50 per cento di probabilità che il bambino risulti portatore del gene e il 25 per cento di probabilità che il bambino sia sano e non portatore della malattia.
By en:User:Cburnett – Own work in Inkscape, CC BY-SA 3.0, Link
I portatori del gene della fibrosi cistica sono sani e non presentano sintomi, quindi possono non rendersi conto della situazione. Spesso i genitori si sentono in colpa nei confronti del figlio affetto da fibrosi cistica, ma è importante ricordare che le cause della fibrosi cistica non sono una diretta conseguenza di comportamenti scelti consapevolmente dai genitori.
Il ruolo degli acidi grassi
Alcuni esperti ritengono che uno squilibrio degli acidi grassi essenziali possa giocare un ruolo importante nella fibrosi cistica. Le persone affette da fibrosi cistica sembrano avere livelli eccessivamente alti di acido arachidonico e una carenza di un altro acido grasso, l’acido docosaexanoico.
Nei portatori sani di un solo gene della fibrosi cistica il livello di acidi grassi è intermedio tra quello dei pazienti affetti da fibrosi cistica e quello dei pazienti che non presentano mutazioni genetiche connesse alla malattia, tuttavia la natura esatta della relazione tra i livelli di acidi grassi e l’anomalia genica che provoca la fibrosi cistica non è chiara.
Fattori di rischio
Il maggior fattore di rischio per la fibrosi cistica sono i precedenti famigliari: se sia voi sia il vostro partner provenite da una famiglia con casi di fibrosi cistica, i vostri figli avranno probabilmente una probabilità su quattro di ammalarsi.
Sintomi
I segni e i sintomi specifici della fibrosi cistica possono essere diversi, a seconda della gravità della malattia. Ad esempio un bambino affetto da fibrosi cistica può avere problemi respiratori ma non digestivi, mentre un altro potrebbe essere interessato da entrambi.
Si assiste inoltre ad un’evoluzione dei sintomi con l’età.
Sintomi nei neonati
In alcuni neonati il primo sintomo può essere un blocco intestinale (ileo da meconio). Si verifica quando il meconio (le feci catramose, di colore nerastro e verdastro che normalmente il neonato produce durante i primissimi uno o due giorni di vita) diventa talmente spesso da ostruire l’intestino. Tra gli altri sintomi nei neonati possiamo avere:
- crescita insufficiente,
- feci abbondanti e oleose (steatorrea),
- infezioni respiratorie frequenti.
Sintomi nei bambini e nei ragazzi
I principali sintomi della fibrosi cistica nei bambini e nei ragazzi comprendono:
- Sapore salato della pelle, perché le persone affette da fibrosi cistica tendono a secernere una quantità di sale (cloruro di sodio) più alta del normale nel sudore. Questo può essere uno dei primi segni che i genitori notano, avvertendo il sapore salato quando baciano il bambino.
- Blocco intestinale.
- Feci oleose, dall’odore forte.
- Ritardo nella crescita.
- Catarro molto viscoso, ma è probabile che i genitori non si accorgano di questo sintomo, perché i bambini tendono a ingoiare il catarro anziché sputarlo.
- Tosse o asma.
- Infezioni frequenti delle alte vie respiratorie e dei seni nasali, complicate da polmonite o bronchite ricorrente.
- Fuoriuscita di parte del retto dall’ano (prolasso rettale), spesso provocata dalla difficoltà nel passaggio delle feci o dalla tosse frequente.
- Rigonfiamento o arrotondamento delle punte delle dita delle mani e dei piedi (dita ippocratiche o a bacchetta di tamburo): nel tempo le dita a bacchetta di tamburo colpiscono la maggior parte delle persone affette da fibrosi cistica, ma compaiono anche in pazienti affetti da malattie cardiache congenite e da problemi polmonari di altro tipo.
La fibrosi cistica può anche essere accompagnata da:
- polipi nasali,
- cirrosi epatica dovuta all’infiammazione o ostruzione dei dotti biliari,
- dislocamento di una parte dell’intestino in una zona diversa (intususcessione), nei bambini di età superiore ai 4 anni.
Pericoli
Complicazioni respiratorie
Tra le complicazioni frequenti della fibrosi cistica troviamo le infezioni croniche delle vie respiratorie, tra cui
- polmonite,
- bronchite,
- sinusite cronica,
- bronchiectasia, una dilatazione anomala delle pareti dei bronchi che rende più difficile la pulizia delle vie respiratorie.
L’asma può essere infine una conseguenza dell’infiammazione cronica delle pareti bronchiali.
Le infezioni delle vie respiratorie sono frequenti perché il muco è più viscoso del normale e per questo ostruisce più facilmente le vie respiratorie, diventando così terreno fertile per proliferazione batterica.
L’agente infettivo che più comunemente interessa i pazienti affette da fibrosi cistica è lo Pseudomonas aeruginosa, un batterio che è causa di aumento delle infezioni dell’apparato respiratorio.
Gli antibiotici possono diminuire la frequenza e la gravità degli attacchi, ma i batteri presenti non vengono mai eradicati completamente dalle vie respiratorie e dai polmoni; d’altra parte questo batterio raramente provoca infezioni polmonari nei soggetti sani e non è comunque considerato contagioso.
I pazienti affetti da fibrosi cistica possono anche soffrire di
- sanguinamento polmonare, che si manifesta attraverso la comparsa di sangue mentre si tossice (emottisi),
- insufficienza respiratoria,
- collasso polmonare (pneumotorace), una malattia in cui l’aria si accumula nella gabbia toracica, fuoriuscendo dal polmone attraverso un piccolo foro che si crea nella parete esterna.
Le malattie polmonari col tempo possono provocare un collasso della parte inferiore destra del cuore (ventricolo destro). Alla fine le complicazioni derivanti da problemi polmonari si rivelano letali per molti pazienti affetti da fibrosi cistica.
Complicazioni nutrizionali
Purtroppo la fibrosi cistica rende i pazienti maggiormente soggetti a diarrea cronica e a gravi carenze nutrizionali.
Le secrezioni viscose ostruiscono i condotti pancreatici, impedendo
- agli enzimi che digeriscono i grassi e le proteine di raggiungere l’intestino,
- all’organismo di assorbire le vitamine liposolubili (A, D, E, K).
La fibrosi cistica colpisce il pancreas e, poiché quest’organo controlla la glicemia (il livello di zucchero nel sangue), il 20% circa dei pazienti sviluppa diabete come conseguenza della malattia.
Infine il dotto biliare (il condotto che porta la bile dal fegato e dalla cistifellea verso l’intestino tenue) può ostruirsi e infiammarsi (colecistite), causando problemi epatici, come la cirrosi.
Complicazioni dell’apparato riproduttivo
La fibrosi cistica colpisce anche l’apparato riproduttivo: spesso le secrezioni viscose bloccano il condotto che collega i testicoli e la prostata (dotto deferente) quindi molti uomini affetti da fibrosi cistica soffrono di sterilità; esistono fortunatamente approcci chirurgici e non in grado di favorire la fertilità, permettendo talvolta agli uomini affetti da fibrosi cistica di diventare padri.
Le donne affette da fibrosi cistica possono essere meno fertili delle altre, però hanno comunque la possibilità di concepire un bambino e di portare a termine la gravidanza con successo. Occorre ricordare, tuttavia, che la gravidanza può far peggiorare i segni e i sintomi della malattia, quindi è essenziale valutare la scelta con lo specialista per soppesare i possibili rischi.
A volte anche l’uso di contraccettivi ormonali (pillola, cerotto, anello) può far peggiorare alcuni sintomi della fibrosi cistica, quindi la scelta va ponderata con l’aiuto del ginecologo.
Quando chiamare il medico
Quando sospettare la presenza di fibrosi cistica?
Andate dal medico immediatamente se temete che vostro figlio sia affetto da fibrosi cistica (anche se in realtà in Italia tutti i bambini dovrebbero essere sottoposti a screening alla nascita).
Tra i sintomi nei neonati possiamo avere:
- disturbi della crescita,
- problemi respiratori cronici (soprattutto polmonite ricorrente),
- frequenti episodi di feci abbondanti e oleose.
I bambini più grandi possono soffrire anche di diarrea e di infezioni respiratorie frequenti, compresa la polmonite. Anche la crescita insufficiente, non in linea con la curva percentile di crescita può essere un sintomo della fibrosi cistica, soprattutto se sono presenti anche altri segni e sintomi.
Quando rivolgersi al medico per complicazioni?
Andate immediatamente dal medico se, dopo la diagnosi, vostro figlio inizia ad avere febbre, tosse che va peggiorando, difficoltà respiratorie, aumento della stanchezza o diminuzione dell’appetito.
Inoltre, se a vostro figlio è stata diagnosticata la fibrosi cistica, programmate esami regolari in una struttura specializzata per la cura di questa malattia. Vostro figlio dovrà anche recarsi regolarmente dal medico di famiglia per farsi prescrivere i farmaci e per tenere sotto controllo gli eventuali problemi respiratori o digestivi.
Diagnosi
Il percorso diagnostico per la fibrosi cistica si è evoluto per garantire una rilevazione sempre più precoce, fondamentale per avviare i trattamenti prima che si verifichino danni d’organo irreversibili.
Screening neonatale
In Italia, lo screening neonatale è obbligatorio e viene eseguito su tutti i neonati nei primi giorni di vita tramite il prelievo di una goccia di sangue dal tallone. Il primo indicatore ricercato è il tripsinogeno immunoreattivo (IRT), una proteina pancreatica che risulta elevata nei neonati con fibrosi cistica. Se i livelli di IRT sono alti, si procede con un’analisi genetica di primo livello per identificare le mutazioni più comuni del gene CFTR.
Il test del sudore
Nonostante i progressi tecnologici, il test del sudore rimane il gold standard per la conferma diagnostica. La procedura prevede la stimolazione della sudorazione su una piccola area del braccio (solitamente tramite pilocarpina e una debole corrente elettrica) e la successiva misurazione della concentrazione di cloruro. Una concentrazione di cloruro superiore a 60 mmol/L è indicativa di fibrosi cistica, mentre valori tra 30 e 59 mmol/L sono considerati “borderline” e richiedono ulteriori approfondimenti.
Analisi genetica e test avanzati
L’analisi del DNA è fondamentale non solo per confermare la diagnosi, ma anche per identificare la specifica mutazione genetica (ne esistono oltre 2.000). Conoscere la mutazione è oggi indispensabile per stabilire l’eleggibilità ai nuovi farmaci modulatori. Nei casi dubbi, possono essere eseguiti test fisiologici avanzati, come la misurazione della differenza di potenziale nasale (NPD) o la misurazione delle correnti intestinali (ICM) su biopsia rettale, che valutano direttamente la funzionalità del canale del cloro.
Cura e terapia
Gli obiettivi del trattamento moderno sono ambiziosi: non si limitano più alla gestione dei sintomi, ma mirano a ripristinare la funzione della proteina difettosa, preservare la funzionalità polmonare e garantire uno stato nutrizionale ottimale. Il piano terapeutico è multidisciplinare e richiede un impegno quotidiano costante.
Modulatori della proteina CFTR
Rappresentano la vera rivoluzione terapeutica degli ultimi anni. Questi farmaci molecolari (come l’associazione elexacaftor/tezacaftor/ivacaftor) agiscono direttamente alla base del difetto biologico, aiutando la proteina CFTR a posizionarsi correttamente sulla membrana cellulare e a funzionare meglio. L’efficacia di questi trattamenti è straordinaria: molti pazienti sperimentano un rapido miglioramento dei test respiratori, una drastica riduzione delle infezioni e un aumento di peso significativo.
Gestione delle complicazioni respiratorie
La pulizia delle vie aeree rimane un pilastro della cura per prevenire il ristagno del muco e le conseguenti infezioni:
- Fisioterapia respiratoria: Tecniche manuali o l’uso di dispositivi a pressione espiratoria positiva (PEP) aiutano a mobilizzare le secrezioni.
- Mucolitici e idratanti: L’inalazione di dornase alfa (un enzima che frammenta il DNA nel muco) o di soluzione salina ipertonica rende le secrezioni più fluide e facili da espellere.
- Terapia antibiotica: Utilizzata per trattare le riacutizzazioni o come mantenimento (spesso per via inalatoria) per controllare la carica batterica cronica, specialmente in presenza di Pseudomonas aeruginosa.
- Broncodilatatori: Somministrati via aerosol per mantenere aperte le vie aeree prima della fisioterapia.
Supporto nutrizionale ed enzimatico
Per contrastare l’insufficienza pancreatica e il malassorbimento, i pazienti devono seguire una strategia nutrizionale rigorosa:
- Enzimi pancreatici: Assunti prima di ogni pasto o spuntino per permettere la digestione di grassi e proteine.
- Integrazione vitaminica: Somministrazione di vitamine liposolubili (A, D, E, K) in formulazioni specifiche.
- Dieta ipercalorica: Spesso è necessario un apporto calorico superiore al 120-150% rispetto alla popolazione generale per sostenere la crescita e la funzione polmonare.
Trapianto d’organo
Quando la malattia polmonare raggiunge uno stadio terminale e le terapie mediche non sono più sufficienti, il trapianto bipolmonare rappresenta un’opzione salvavita. Sebbene non curi la fibrosi cistica negli altri organi, il trapianto può offrire molti anni di vita con una funzione respiratoria normalizzata.
Stile di vita e gestione quotidiana
L’attività fisica regolare è considerata a tutti gli effetti parte della terapia: lo sport aiuta la clearance del muco, migliora la capacità aerobica e ha effetti positivi sull’umore e sulla densità ossea. È essenziale mantenere un’idratazione abbondante e, durante i mesi estivi o l’attività intensa, integrare i sali minerali persi con il sudore.
I pazienti e le famiglie devono prestare particolare attenzione alle misure igieniche (lavaggio frequente delle mani) e alla prevenzione delle infezioni crociate, evitando il contatto stretto con altri pazienti affetti da fibrosi cistica per prevenire lo scambio di batteri multiresistenti.
La ricerca sulla terapia genica e sull’editing genomico (CRISPR/Cas9) prosegue intensamente con l’obiettivo futuro di correggere definitivamente il difetto genetico nelle cellule staminali del polmone, una prospettiva che potrebbe un giorno portare alla guarigione completa.
Fonti e bibliografia
Le domande più frequenti
Cos'è la fibrosi cistica?
Quali sono i sintomi? Quando sospettare la presenza di fibrosi cistica?
Come si cura?
Tutti gli aggiornamenti su salute, alimentazione e benessere.