Introduzione
L’insonnia familiare fatale è una malattia neurodegenerativa ereditaria molto rara e purtroppo invariabilmente fatale. La malattia è infatti attualmente incurabile e presenta un decorso medio di 18 mesi (in letteratura di descrivono casi dai 7 ai 36 mesi), che alla fine porta alla morte.
È caratterizzata dall’incapacità di dormire, inizialmente in forma lieve, ma con un progressivo peggioramento fino a determinare un significativo deterioramento fisico e mentale.
La malattia fu descritta per la prima volta nel 1765, peraltro in un paziente italiano, sebbene sia stata poi formalmente descritta e riconosciuta clinicamente nel 1986 da Lugaresi ed altri.

Shutterstock/Gorodenkoff
Cause
L’insonnia familiare fatale è una malattia da prioni, ovvero causata dalla degenerazione di una proteina normale in una forma differente, non più solubile in acqua e resistente ai tentativi di degradazione cellulare, con il conseguente progressivo e inevitabile accumulo a livello del sistema nervoso centrale. Una malattia che colpisce allo stesso modo, divenuta piuttosto nota qualche anno fa, è la malattia di Creutzfeldt-Jakob (tristemente famosa nella sua forma denominata morbo della mucca pazza).
Nel caso dell’insonnia familiare fatale la parte più colpita dall’accumulo risulta il talamo, una struttura del cervello.
L’origine di questa modificazione è genetica, con trasmissione secondo il modello autosomico dominante:
- autosomico: la mutazione si trova su un cromosoma di tipo non sessuale (il numero 20),
- dominante: è sufficiente che la mutazione sia presente su un’unica copia del cromosoma (il gene anomalo può cioè derivare da un solo genitore).
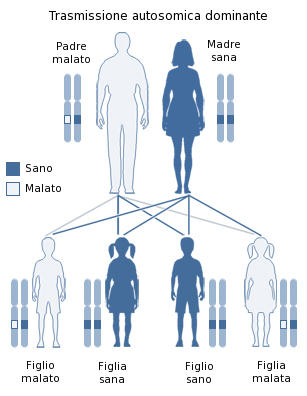
Modello di trasmissione genetica autosomico-dominante (Di LoStrangolatore – Opera propria, CC0, https://commons.wikimedia.org/w/index.php?curid=17919448)
Il rischio di trasmettere il gene anomalo dal genitore affetto alla prole è del 50% per ogni gravidanza, indipendentemente dal sesso del nascituro; questo modello di trasmissione non prevede l’esistenza di portatori sani.
Le malattie genetiche da prioni sono molto rare e l’insonnia familiare fatale è particolarmente rara (ad oggi la mutazione che causa la malattia è stata descritta in circa 50 famiglie in tutto il mondo).
In rari casi la mutazione responsabile della malattia si verifica spontaneamente, senza una storia familiare della condizione (de novo). La variazione genetica si verifica in questo caso al momento della formazione dell’ovulo o dello spermatozoo, senza che nessun membro della famiglia ne fosse già affetto, ma il paziente diventerà invece capace di trasmettere il gene mutato alla propria prole in modo autosomico dominante.
Il gene colpito dalla mutazione (PRNP) è responsabile della produzione della proteina prionica umana, la cui funzione fisiologica non è ad oggi nota.
Sintomi
I pazienti affetti da insonnia familiare fatale mostrano l’insorgenza dei sintomi tra i 20 e i 60 anni circa, senza differenze tra uomini e donne.
I soggetti colpiti inizialmente lamentano una generica insonnia (ovvero una riduzione del tempo totale di sonno), che aumenta di gravità con il progredire della malattia. Piuttosto comune la successiva sonnolenza diurna, così come l’esperienza di sogni lucidi (ovvero vissuti con la consapevolezza di stare dormendo, da cui deriva la capacità di muoversi in modo deliberato) nonostante le limitate ore di riposo.
l paziente può presentare vari segni obiettivi di coinvolgimento neurologico, come:
- disfunzioni del sistema nervoso autonomo (la parte che controlla i processi corporei involontari):
- pressione alta,
- episodi di tachipnea (aumento della frequenza respiratoria),
- aumento della lacrimazione e/o sudorazione,
- costipazione,
- disfunzioni sessuali,
- alterazioni della temperatura corporea,
- visione doppia,
- disartria (disturbi del linguaggio),
- difficoltà di deglutizione,
- anomalie dello sguardo,
- rallentamento dell’elaborazione del pensiero,
- disturbi dell’attenzione,
- perdita di memoria a breve termine,
- atassia (perdita di coordinazione),
- perdita di peso,
Con il progredire della malattia il paziente sviluppa uno stato simile al delirio, sebbene le capacità comportamentali e intellettuali tendano ad essere preservate anche nelle ultime fasi della malattia.
I cambiamenti di umore sono comuni, tipicamente con sviluppo di depressione e/o apatia con il peggioramento dell’insonnia.
Si possono osservare cambiamenti nel tono muscolare associati a debolezza e movimenti anormali (anche questi tendono a peggiorare con la durata della malattia).
Decorso
L’insonnia familiare fatale può essere didatticamente distinta in quattro fasi successive:
- Il primo stadio della malattia è caratterizzato dall’insorgenza dell’insonnia, che peggiora nell’arco di pochi mesi e provoca sintomi psichiatrici come fobia, paranoia e attacchi di panico. È durante questo stadio che i pazienti possono riferire di vivere sogni lucidi.
- Nel successivo periodo di 5 mesi, i sintomi psichiatrici peggiorano insieme con l’insonnia e compaiono allucinazioni. Si osserva disfunzione autonomica (aumento della pressione del sangue, si sviluppano episodi di tachipnea, aumento di lacrimazione/sudorazione, stitichezza, …).
- La terza fase, della durata di circa 3 mesi, è tipicamente dominata da un’insonnia ormai assoluta.
- La fase finale della malattia può durare sei mesi o più ed è definita da rapido declino cognitivo e compara di demenza. I pazienti perdono la capacità di muoversi o parlare volontariamente.
Il paziente va infine incontro a coma e successiva morte.
Diagnosi
La diagnosi di insonnia familiare fatale richiede la valutazione di una combinazione di valutazione clinica (osservazione dei sintomi caratteristici), anamnestica (raccolta di informazioni relative a storia clinica, familiarità, …) ed esami strumentali.
I test genetici molecolari possono confermare la diagnosi in alcuni casi, mentre la polisonnografia e la PET sono utili per dimostrare rispettivamente la riduzione del tempo di sonno effettivo e una riduzione dell’attività cerebrale a livello del talamo.
Altre tecniche di imaging, come la tomografia computerizzata (TC) e la risonanza magnetica, possono contribuire all’esclusione di altre condizioni che entrano in diagnosi differenziale con l’insonnia familiare fatale.
Il RTQuIC è infine un test di recente introduzione in grado di valutare la presenza di di prioni nel liquido cerebrospinale.
Cura
La terapia è volta principalmente a fornire un sollievo ai sintomi e su cure palliative, perché purtroppo ad oggi non esiste cura per l’insonnia familiare fatale. A peggiorare la situazione è la rarità della condizione, che ne limita tanto la ricerca quanto l’esperienza clinica.
È stato osservato che:
- è di beneficio per il paziente interrompere eventuli farmaci in grado di peggiorare i sintomi (confusione, memoria, insonnia, …)
- non si manifesta una risposta soddisfacente ai sedativi in termini di tracciato dell’elettroencefalogramma, a prescindere dalla classe di farmaci usati (barbiturici, benzodiazepine), mentre è stato aneddoticamente di beneficio la somministrazione di gamma-idrossibutirrato
- nelle fasi avanzate potrebbe rendersi necessaria l’applicazione di un sondino per l’alimentazione (in caso di difficoltà di deglutizione).
La terapia psicosociale è importante sia per il paziente che per la famiglia, che nelle fasi terminali può beneficiare del supporto di un hospice.
Fonti e bibliografia
- Fatal Familial Insomnia – Zalan Khan; Pradeep C. Bollu
- RareDiseases
Tutti gli aggiornamenti su salute, alimentazione e benessere.






