Introduzione
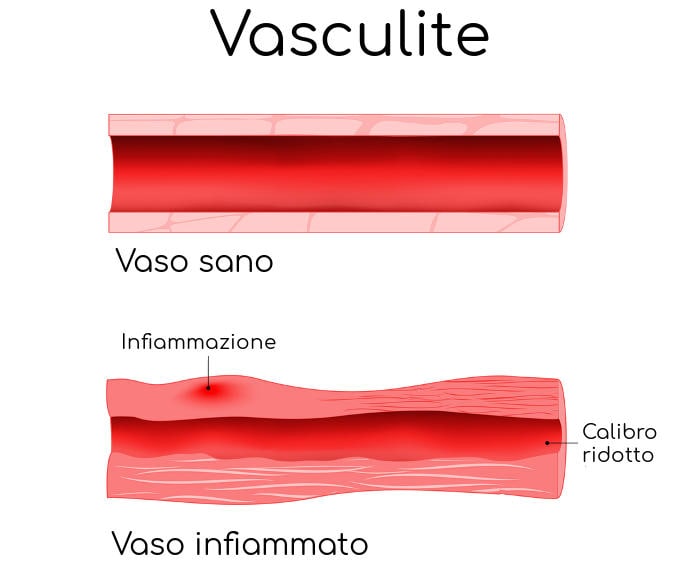
La Sindrome di Behcet è una malattia sistemica piuttosto rara; si tratta di una vasculite che colpisce i vasi sanguigni di piccolo e medio calibro, portando ad alterazioni patologiche di diversi organi ed apparati.
L’eziologia è ancora incerta, ma si ritiene che ci sia alla base una reazione autoimmune, ovvero un’attivazione del sistema immunitario verso cellule del proprio organismo in seguito ad alcune disregolazioni genetiche ed acquisite.
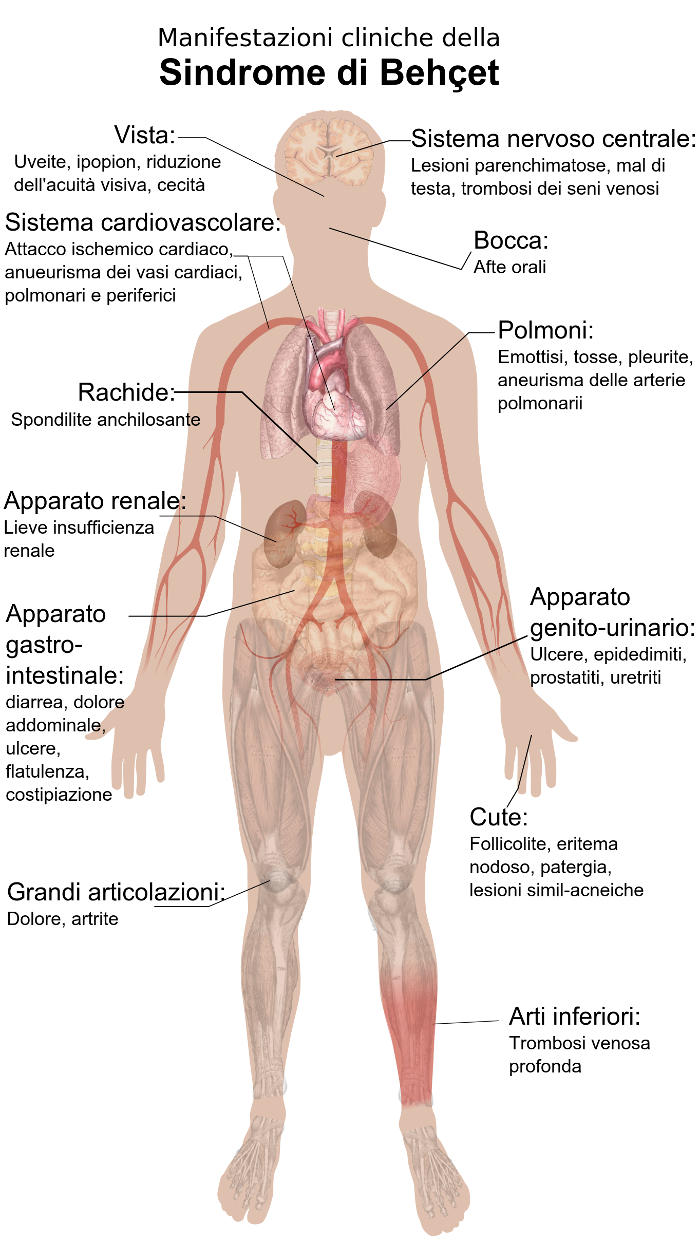
La sindrome di Behcet può interessare differenti distretti corporei, ma generalmente si manifesta con la comparsa di 3 sintomi principali:
- afte orali (ulcerazioni a livello di lingua, gengive, palato e in genere del cavo orale),
- afte genitali,
- uveite (infiammazione della tonaca vascolare dell’occhio),
ma molto comuni sono anche i sintomi pseudoinfluenzali come:
- astenia,
- malessere,
- febbre
- e dolori muscolari ed articolari.
Tra gli altri possibili sintomi ricordiamo quelli in grado di interessare i sistemi
- gastro-intestinale,
- cardiovascolare,
- cutaneo.
La diagnosi è principalmente di tipo clinico, basato cioè sull’osservazione dei principali segni oggettivi e sintomi soggettivi del paziente. È sempre necessaria una minuziosa diagnosi differenziale prima di poter intraprendere un trattamento mirato ed efficace.
La cura per la sindrome di Behcet è oggi basata su un approccio multidisciplinare che utilizza farmaci antinfiammatori, immunosoppressori e terapie biologiche di ultima generazione. Questo tipo di trattamento permette di alleviare notevolmente i sintomi, prevenire i danni d’organo e migliorare significativamente la qualità della vita. È richiesto in ogni caso uno stretto follow-up nel corso degli anni per gestire al meglio la presenza dei sintomi e per eventuali adeguamenti della terapia.
Di Adert – Opera propria, CC BY-SA 3.0, Collegamento
Cause
La malattia di Behcet è una patologia
- autoimmune (il sistema immunitario si attiva contro l’organismo stesso)
- sistemica (che può potenzialmente coinvolgere l’intero organismo)
- cronica (persistente nel tempo e non destinata a risolversi),
che interessa la popolazione giovanile, manifestandosi principalmente
- tra i 20 e i 50 anni,
- soprattutto nel sesso femminile (dove l’incidenza di patologie autoimmuni è quasi sempre superiore rispetto al sesso maschile).
La patogenesi è basata su alterazioni genetiche che prevedono la presenza di alcuni antigeni cellulari come l’HLA-B51 e l’HLA-B57. La presenza di questi antigeni aumenta notevolmente il rischio che i soggetti portatori possano sviluppare la malattia nel corso degli anni.
Semplificando il concetto, si verificano degli errori di funzionamento del sistema immunitario che, anziché colpire i germi patogeni, riconosce come patogene alcune strutture “self” come i vasi sanguigni (soprattutto arteriosi) di piccolo e medio calibro. Questo porta allo sviluppo di un processo infiammatorio cronico che a lungo andare porta alla manifestazioni dei sintomi caratteristici della condizione.
A favorire questa disregolazione del sistema immunitario può contribuire un altro fattore: normalmente gli antigeni di vari microrganismi patogeni vengono riconosciuti come estranei e pericolosi, venendo perciò attaccati da diverse cellule immunitarie. Nei soggetti affetti dalla Behcet può capitare che questi antigeni estranei posseggano una struttura molecolare molto simile ad antigeni “self” del nostro organismo. Per questa ragione le cellule immunitarie come neutrofili e linfociti attaccano gli antigeni “self” scambiandoli per antigeni estranei, portando allo sviluppo di una reazione infiammatoria autoimmune.
Sintomi
Essendo una malattia sistemica, la sindrome di Behcet può colpire qualsiasi organo ed apparato con numerosi e variegati sintomi; nella maggior parte dei casi i sintomi preponderanti sono:
- afte orali,
- afte genitali,
- uveite.
Per afta si intende una piccola ulcera a livello di una mucosa che si manifesta con
- bruciore,
- arrossamento,
- dolore urente.
A livello orale le afte sono molto frequenti e vengono chiamate in gergo “bolle”.
In caso di sindrome di Behcet le afte orali si presentano molto più frequentemente del normale e sono multiple, comparendo contemporaneamente in diversi punti del cavo orale. Anche la frequenza con cui tendono a comparire è nettamente maggiore.
Nella sindrome di Behcet queste afte si presentano anche a livello genitale a livello di
- pene e/o glande nell’uomo,
- piccole e grandi labbra nella donna.
Per uveite invece si intende l’infiammazione dell’uvea, ovvero la tonaca vascolare dell’occhio. In caso di uveite compaiono i tipici occhi “rossi” associati a
- dolore,
- disturbo della visione,
- lacrimazione
- e fotofobia (senso di fastidio agli occhi provocato dalla luce di intensità normale).
Oltre a questa tipica triade, i sintomi della malattia di Behcet possono presentarsi a carico di:
- Sistema gastro-intestinale:
- dolore addominale,
- meteorismo,
- ulcere gastriche, intestinali o anali.
- Apparato urinario:
- il rene può essere interessato da glomerulonefriti o da amiloidosi con rischio medio-basso di sviluppare insufficienza renale.
- Sistema muscolo-scheletrico:
- dolori a diverse articolazioni, soprattutto a livello di ginocchio, gomito, polso e anca. Spesso l’artrite diventa un sintomo molto invalidante, tanto da richiedere un approfondimento diagnostico ed una terapia efficace.
- Sistema cardiovascolare:
- piuttosto pericolosa può risultare la formazione di aneurismi a livello dei vasi arteriosi come l’aorta o di arterie cerebrali (con rischio di dissezione aortica, ictus o emorragie cerebrali). A livello del cuore possono presentarsi endocardite, miocardite e pericardite.
- Apparato cutaneo:
- eritemi,
- vescicole
- o nodulazioni.
Infine la malattia di Behcet si presenta con sintomi di tipo pseudo-influenzale come febbre più o meno elevata e persistente per diversi giorni a settimana, astenia e malessere generalizzato.
Diagnosi
La diagnosi della sindrome di Behçet è una sfida clinica, poiché non esiste un singolo esame di laboratorio o radiologico che possa confermarla con certezza assoluta. Il medico giunge alla conclusione basandosi principalmente sulla storia clinica del paziente e sulla presenza di criteri standardizzati a livello internazionale.
Criteri diagnostici internazionali
Attualmente, i medici utilizzano i criteri dell’International Criteria for Behçet’s Disease (ICBD), un sistema a punti che attribuisce un valore specifico alle diverse manifestazioni della malattia:
- Afte orali ricorrenti: (2 punti) Definitive e persistenti, devono presentarsi almeno tre volte in un anno.
- Afte genitali: (2 punti) Ulcere spesso più profonde e dolorose di quelle orali.
- Lesioni oculari: (2 punti) Come l’uveite anteriore o posteriore e la vasculite retinica.
- Lesioni cutanee: (1 punto) Inclusi l’eritema nodoso o lesioni simili all’acne (pseudofollicolite).
- Manifestazioni vascolari: (1 punto) Presenza di trombosi venose o aneurismi arteriosi.
- Manifestazioni neurologiche: (1 punto) Sintomi legati al coinvolgimento del sistema nervoso centrale.
- Test della patergia positivo: (1 punto addizionale) Un test cutaneo specifico.
Una diagnosi di Behçet è generalmente considerata certa quando il paziente totalizza un punteggio pari o superiore a 4 punti.
Test della patergia e analisi strumentali
Il test della patergia è uno strumento diagnostico molto utile: consiste in una piccola puntura sull’avambraccio con un ago sterile. Se dopo 24-48 ore si forma una piccola protuberanza rossa o una pustola, il test è positivo, indicando un’iper-reattività del sistema immunitario a traumati minimi. Sebbene sia molto specifico per la sindrome di Behçet, molti pazienti (specialmente in Europa) possono risultare negativi pur essendo affetti dalla patologia.
Ulteriori esami sono necessari per escludere altre malattie e valutare il coinvolgimento degli organi interni:
- Esami del sangue: Si ricercano i marker dell’infiammazione (VES e Proteina C Reattiva) e l’antigene HLA-B51, che pur non essendo diagnostico di per sé, supporta il sospetto clinico.
- Imaging: La Risonanza Magnetica (RM) è fondamentale se si sospetta un Neuro-Behçet, mentre l’angio-TC o l’ecografia Doppler sono utilizzate per individuare eventuali complicanze vascolari.
- Esame oculistico: Fondamentale per identificare precocemente segni di uveite che potrebbero non essere ancora sintomatici.
È essenziale una diagnosi differenziale accurata per distinguere il Behçet da altre vasculiti, malattie infiammatorie intestinali (come il morbo di Crohn) o infezioni virali.
Cura
Il trattamento della sindrome di Behçet ha come obiettivi principali il controllo rapido delle fasi acute (flare), la prevenzione delle recidive e la protezione degli organi vitali da danni permanenti. Poiché la malattia si manifesta in modo diverso da persona a persona, la terapia è strettamente personalizzata e adattata alla gravità e alla localizzazione delle infiammazioni.
Opzioni terapeutiche principali
Le strategie di cura si dividono generalmente in trattamenti locali per le manifestazioni lievi e terapie sistemiche per le forme più complesse:
- Trattamenti topici: Per le afte orali e genitali si utilizzano collutori, creme o gel a base di corticosteroidi. Questi aiutano a ridurre il dolore e accelerare la guarigione delle ulcere.
- Colchicina: È spesso il farmaco di prima scelta, specialmente per prevenire la ricorrenza delle afte e per gestire i dolori articolari e le lesioni cutanee.
- Corticosteroidi: Farmaci come il prednisone sono fondamentali per spegnere l’infiammazione durante le fasi acute. Possono essere somministrati per via orale o, nei casi gravi (come il coinvolgimento oculare o neurologico), tramite infusioni endovenose ad alto dosaggio.
- Immunosoppressori: Se i corticosteroidi non sono sufficienti o se si rende necessario ridurne il dosaggio per evitare effetti collaterali, vengono introdotti farmaci come l’azatioprina, la ciclosporina o il metotrexato. Questi farmaci “calmano” il sistema immunitario nel lungo periodo.
Terapie biologiche e innovative
Negli ultimi anni, la gestione della sindrome di Behçet è stata rivoluzionata dai farmaci biologici, che colpiscono in modo mirato specifiche molecole del sistema immunitario responsabili dell’infiammazione:
- Inibitori del TNF-alfa: (come infliximab e adalimumab) Sono estremamente efficaci nelle forme gravi di uveite, nel coinvolgimento del sistema nervoso e nelle ulcere gastrointestinali resistenti.
- Apremilast: Un farmaco orale recentemente approvato specificamente per il trattamento delle afte orali resistenti in pazienti con Behçet.
- Inibitori dell’interleuchina: Nuove opzioni terapeutiche vengono utilizzate in casi selezionati quando le altre terapie non sortiscono gli effetti sperati.
Stile di vita e raccomandazioni pratiche
Sebbene la terapia farmacologica sia il pilastro della cura, alcuni accorgimenti quotidiani possono fare la differenza nella gestione della malattia:
- Igiene orale rigorosa: L’uso di spazzolini a setole morbide e il controllo regolare dal dentista aiutano a ridurre l’irritazione che può scatenare le afte orali.
- Alimentazione: Durante le fasi di stomatite acuta, è consigliabile evitare cibi troppo acidi, salati, piccanti o molto caldi che possono esacerbare il dolore delle ulcere.
- Gestione dello stress e riposo: Molti pazienti riferiscono che periodi di forte stress o stanchezza intensa possono scatenare un peggioramento dei sintomi. Un riposo adeguato e tecniche di rilassamento sono parte integrante del benessere complessivo.
- Esercizio fisico: Un’attività fisica moderata è raccomandata per mantenere la mobilità articolare e il benessere cardiovascolare, avendo cura di riposare durante le fasi di artrite acuta.
La prognosi è oggi molto più favorevole rispetto al passato grazie alla diagnosi precoce e alla disponibilità di farmaci biologici, che permettono a molti pazienti di condurre una vita attiva e produttiva.
Fonti e bibliografia
- Harrison – Principi Di Medicina Interna Vol. 1 (17 Ed. McGraw Hill 2009)
- Linee Guida EULAR (European Alliance of Associations for Rheumatology) per la gestione della malattia di Behçet.
Tutti gli aggiornamenti su salute, alimentazione e benessere.