Introduzione
La sclerodermia è una malattia cronica autoimmune che causa infiammazione e fibrosi (ispessimento) nella pelle e in altre aree del corpo (il nome della condizione deriva dal greco antico “pelle dura”).
È considerata una malattia rara ed è scatenata da un errore di riconoscimento commesso dal sistema immunitario, che si attiva innescando una reazione infiammatoria diretta verso tessuti sani; come conseguenza il corpo produce una quantità eccessiva di collagene, la proteina responsabile della formazione del tessuto connettivo.
Questo eccesso di collagene si accumula nella pelle e in altri tessuti, provocando la formazione di aree cutanee tese e indurite, caratteristiche della sclerodermia.
Purtroppo la deposizione di collagene non si limita alla pelle, ma può coinvolgere molteplici sistemi dell’organismo, rendendo la sclerodermia una patologia sistemica potenzialmente grave, che nelle forme più gravi si manifesta con una progressiva sostituzione dei normali tessuti degli organi interni (polmoni, esofago, vasi sanguigni, cuore, reni) attraverso un processo di fibrosi.
La complessità della sclerodermia richiede un approccio terapeutico multidisciplinare, che può includere trattamenti farmacologici, fisioterapia e, in alcuni casi, terapie infiltrative mirate a ridurre l’infiammazione e migliorare la flessibilità dei tessuti colpiti.

Sclerodermia localizzata sulla schiena, di Leith C Jones di Wikipedia in inglese, CC BY 3.0, Collegamento
Cause
Esistono due tipi principali di sclerodermia:
- Sclerodermia localizzata: è una forma benigna, limitata alla pelle e/o al tessuto sottocutaneo. Può essere a sua volta distinta in due forme:
- lineare, quando le lesioni della pelle sono di modesta entità e hanno un aspetto ben definito;
- morfea, cioè aree di pelle indurita a placche.
- Sclerodermia sistemica (o sclerosi sistemica), condizione più grave caratterizzata da indurimento della pelle e coinvolgimento degli organi interni. Può essere:
- limitata, se le lesioni ispessite della pelle si riscontrano solo a livello delle braccia, delle gambe e delle dita della mano e dei piedi;
- diffusa, quando riguarda anche il viso, la regione ascellare, il torace e il dorso, fino ad avere manifestazioni di danno d’organo dovute all’alterazione della circolazione nei vasi sanguigni. Nelle forme avanzate è una patologia molto invalidante che peggiora sensibilmente la qualità di vita dei soggetti affetti.
L’esatta causa dell’insorgenza della sclerodermia è tuttora sconosciuta, ma si ritiene che possa derivare dalla combinazione di fattori genetici e ambientali:
- La sclerosi sistemica si presenta spesso con una certa familiarità, eventualmente associata ad altre malattie autoimmuni;
- diversi fattori scatenanti ambientali sono associati allo sviluppo successivo della sclerosi sistemica, tra cui agenti infettivi come il citomegalovirus (CMV), il virus di Epstein-Barr e il parvovirus B19. Anche l’esposizione alla polvere di silice è stata associata alla sclerosi sistemica, insieme all’esposizione occasionale ad altri agenti chimici. Ad oggi, invece, il fumo di sigaretta non sembra essere un fattore di rischio.
La fibrosi
Dal punto di vista fisiopatologico la sclerodermia provoca infiammazione e fibrosi (un processo patologico caratterizzato dall’accumulo di tessuto connettivo e cicatriziale) nella pelle e in altri organi del corpo.
Nel contesto della sclerodermia, la fibrosi si verifica a seguito di un errore del sistema immunitario, che attacca erroneamente i tessuti sani come se fossero corpi estranei. Questo innesca una risposta infiammatoria prolungata, che stimola i fibroblasti (cellule responsabili della produzione del collagene) a produrre una quantità eccessiva di collagene, la principale proteina strutturale del tessuto connettivo.
Il collagene, in condizioni normali, ha la funzione di fornire struttura e forza a pelle, tendini, vasi sanguigni e altri organi. Tuttavia, quando la produzione di collagene diventa incontrollata, si accumula nei tessuti in modo anomalo. Questo provoca l’ispessimento e l’indurimento della pelle e, in alcune forme di sclerodermia, anche degli organi interni.
Il processo di fibrosi non si limita alla pelle: l’eccesso di collagene può sostituire gradualmente i tessuti normali degli organi interni, compromettendone la funzione. Ad esempio, nei polmoni, la fibrosi può interferire con la capacità respiratoria, mentre nei reni può portare a insufficienza renale. Questo rende la sclerodermia una malattia sistemica potenzialmente grave e altamente debilitante, specialmente nelle forme più avanzate.
Fattori di rischio
La sclerodermia non è considerata una malattia ereditaria, tuttavia è stata riconosciuta la familiarità con altre patologie autoimmuni: diversi soggetti affetti da sclerodermia presentano familiari con altre malattie autoimmuni, come per esempio l’artrite reumatoide o il lupus eritematoso sistemico.
- La sclerodermia è più comune nelle donne che negli uomini.
- La malattia di solito si manifesta tra i 30 e i 50 anni ed è più comune negli adulti che nei bambini.
Sintomi
I sintomi della sclerodermia variano da persona a persona, soprattutto in base alla forma di cui si soffre:
- Sclerodermia localizzata, che in genere causa chiazze di pelle spessa e dura con caratteristiche peculiari:
- La sclerodermia morfea si manifesta con l’ispessimento di chiazze di pelle di forma ovale. Queste aree possono avere un aspetto giallo e ceroso circondato da un bordo rossastro o simile a un livido. Le chiazze possono rimanere confinate in una sola area, o diffondersi in altri distretti. Alcuni pazienti lamentano anche una sensazione di affaticamento e stanchezza.
- La sclerodermia lineare provoca la formazione di linee di pelle ispessita o di colore diverso che corrono lungo il braccio, la gamba e, più raramente, sulla fronte.
- La sclerodermia sistemica (scleroderma) può avere un esordio rapido e improvviso o più graduale, ma soprattutto è in grado di causare alterazioni agli organi interni oltre che alla pelle. È comune la sensazione di affaticamento.
- La sclerodermia cutanea limitata si manifesta gradualmente e di solito colpisce la pelle di dita, mani, viso, avambracci e gambe sotto le ginocchia. Spesso arriva a causare problemi ai vasi sanguigni e all’esofago (con difficoltà a deglutire). La forma limitata ha un coinvolgimento meno frequente degli organi interni importanti, come malattie renali o malattie polmonari progressive; tende a peggiorare gradualmente nel tempo, sebbene sia generalmente meno grave della sclerosi sistemica diffusa e possa essere efficacemente controllata con un’adeguata terapia.
- La sclerodermia cutanea diffusa si manifesta all’improvviso, di solito con ispessimento della pelle sulle dita delle mani o dei piedi. L’ispessimento della pelle si diffonde poi al resto del corpo sopra i gomiti e/o le ginocchia. Questo tipo può anche danneggiare gli organi interni (intero apparato digerente, polmoni, reni, cuore, con il rischio di sviluppare mancanza di respiro, pressione alta e ipertensione polmonare) ed essere accompagnata dasintomi sistemici come perdita di peso, affaticamento, dolori e rigidità alle articolazioni.
Comorbidità
Altre manifestazioni della sclerosi sistemica includono l’ipotiroidismo, che colpisce fino al 15% dei pazienti, e altri disturbi tiroidei come malattie autoimmuni della tiroide, come la tiroidite di Hashimoto e la malattia di Graves. Questi pazienti presentano anche un rischio maggiore di sviluppare altre condizioni autoimmuni, come la cirrosi biliare primitiva e la sindrome di Sjögren.
I pazienti con sclerosi sistemica sono stati associati a un aumentato rischio di sviluppare disturbi psicologici, come la depressione, che può colpire fino al 50% dei soggetti.
Fenomeno di Raynaud
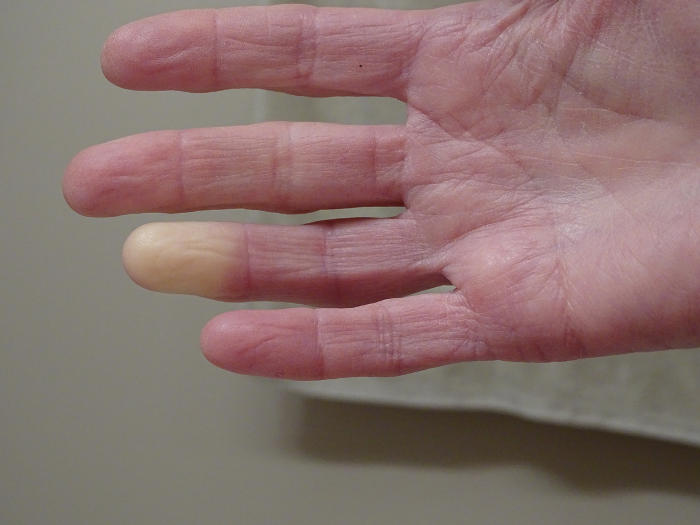
Fenomeno di Raynaud, iStock.com/arb Elkin
Il fenomeno di Raynaud è una manifestazione precoce comune, osservata in oltre il 95% dei pazienti con sclerosi sistemica.
La condizione comporta vasospasmi innescati dall’esposizione al freddo, che determinano cambiamenti di colore successivi in 3 fasi, che in genere interessano le dita, in particolare nelle estremità superiori:
- pallore iniziale con scolorimento bianco ben definito,
- seguito da cambiamenti ischemici con un aspetto bluastro scuro
- e, infine, iperemia reattiva che porta a scolorimento rosso
Il fenomeno di Raynaud può manifestarsi anche anni prima dell’insorgenza del coinvolgimento viscerale, in particolare nei casi di sclerosi sistemica cutanea limitata, ma è importante notare che fino al 15% delle persone sperimenta il fenomeno di Raynaud senza mai sviluppare la progressione verso la sclerodermia.
Diagnosi
La diagnosi di sclerodermia, e in particolare della sclerosi sistemica, è un processo complesso che richiede l’integrazione di segni clinici, test di laboratorio e indagini strumentali avanzate. L’obiettivo principale dei protocolli attuali è la diagnosi precoce, fondamentale per intervenire prima che il danno agli organi interni diventi irreversibile.
Il sospetto clinico nasce solitamente in presenza di tre elementi chiave: il fenomeno di Raynaud, l’ispessimento cutaneo (spesso valutato mediante il punteggio “Rodnan skin score” che misura la plicabilità della pelle in 17 aree del corpo) e il gonfiore delle dita (“puffy fingers”).
Esami di laboratorio e biomarcatori
Gli esami del sangue sono essenziali per confermare l’origine autoimmune della patologia. Vengono ricercati anticorpi specifici che non solo aiutano nella diagnosi, ma offrono anche indicazioni sulla possibile evoluzione della malattia:
- Anticorpi Antinucleo (ANA): Positivi nella quasi totalità dei pazienti (oltre il 95%).
- Anticorpi anti-centromero (ACA): Tipicamente associati alla forma cutanea limitata e a una progressione più lenta.
- Anticorpi anti-Scl-70 (anti-topoisomerasi I): Suggeriscono spesso la forma cutanea diffusa e un rischio maggiore di coinvolgimento polmonare.
- Anticorpi anti-RNA polimerasi III: Associati a un rapido coinvolgimento cutaneo e a un rischio aumentato di crisi renale sclerodermica.
Indagini strumentali
Per definire l’estensione della malattia e monitorare gli organi interni, si ricorre a diversi esami:
- Videocapillaroscopia periungueale: È l’esame d’elezione per lo screening precoce. Attraverso una sonda ottica, il medico osserva i capillari alla base dell’unghia; la presenza di capillari dilatati (megacapillari) o aree prive di vasi (aree avascolari) è un segno distintivo della malattia.
- Screening polmonare: Include la TAC del torace ad alta risoluzione (HRCT) per individuare precocemente la fibrosi interstiziale e le prove di funzionalità respiratoria con test della diffusione del monossido di carbonio (DLCO).
- Valutazione cardiaca: L’ecocardiogramma color-doppler è fondamentale per monitorare la pressione nelle arterie polmonari e prevenire l’ipertensione polmonare.
- Monitoraggio gastrointestinale: In presenza di sintomi, possono essere necessarie la manometria esofagea o l’endoscopia per valutare la motilità e l’eventuale danno da reflusso.
Cura
Attualmente non esiste una cura risolutiva per la sclerodermia, ma i progressi terapeutici degli ultimi anni hanno trasformato radicalmente la prognosi e la qualità della vita dei pazienti. Il trattamento moderno è “organo-specifico”: non si cura solo la malattia nel suo insieme, ma si interviene in modo mirato su ogni distretto colpito (pelle, polmoni, cuore, reni, apparato digerente).
Gli obiettivi della terapia sono spegnere l’infiammazione, rallentare il processo di fibrosi e gestire le complicanze vascolari.
Gestione dei problemi vascolari
Per contrastare il fenomeno di Raynaud e prevenire le ulcere digitali (piccole ferite dolorose sulla punta delle dita causate dalla mancanza di sangue), si utilizzano farmaci che dilatano i vasi sanguigni:
- Calcio-antagonisti: Sono la prima scelta per ridurre la frequenza e l’intensità dei vasospasmi.
- Vasodilatatori potenti: In casi più severi si ricorre a farmaci come i prostanoidi (es. iloprost), somministrati solitamente per via endovenosa in cicli periodici, o antagonisti dei recettori dell’endotelina (es. bosentan).
- Inibitori della fosfodiesterasi-5: Farmaci come il sildenafil possono essere impiegati per migliorare il flusso sanguigno periferico.
Terapia immunosoppressiva e biologica
Per modulare la risposta del sistema immunitario e rallentare l’ispessimento cutaneo e polmonare, il medico può prescrivere:
- Micofenolato mofetile: Attualmente considerato uno dei farmaci di riferimento per il trattamento della fibrosi polmonare e cutanea.
- Metotrexato: Utilizzato prevalentemente nelle forme con importante coinvolgimento della pelle e delle articolazioni.
- Corticosteroidi: Come il prednisone, vengono usati con estrema cautela e a bassi dosaggi, poiché dosi elevate possono aumentare il rischio di crisi renali.
- Anticorpi monoclonali: Il rituximab e il tocilizumab hanno mostrato benefici significativi nel controllo della malattia polmonare e della progressione cutanea in casi selezionati.
- Nintedanib: Un farmaco anti-fibrotico specifico approvato per rallentare il declino della funzione polmonare nei pazienti con fibrosi interstiziale associata a sclerosi sistemica.
Gestione delle complicanze d’organo
- Apparato digerente: Si utilizzano inibitori di pompa protonica per il reflusso e procinetici per migliorare la motilità esofagea e intestinale.
- Reni: L’uso tempestivo degli ACE-inibitori ha rivoluzionato il trattamento della crisi renale sclerodermica, un tempo spesso fatale.
- Trapianto di cellule staminali: In casi selezionati di malattia diffusa a rapida progressione e senza gravi danni d’organo preesistenti, il trapianto autologo di cellule staminali emopoietiche può essere preso in considerazione per “resettare” il sistema immunitario.
Stile di vita e fisioterapia
Il ruolo del paziente è centrale nella gestione quotidiana della malattia. La protezione dal freddo è imperativa: l’uso di guanti termici, scaldini e indumenti stratificati può prevenire gli attacchi di Raynaud. La cura della pelle richiede l’applicazione costante di creme emollienti per mantenere l’elasticità e ridurre il prurito.
La fisioterapia e la terapia occupazionale sono fondamentali per preservare la mobilità articolare delle mani e del viso (esercizi di mimica facciale). Dal punto di vista nutrizionale, si consigliano pasti piccoli e frequenti per gestire i disturbi della deglutizione e il reflusso, evitando di coricarsi immediatamente dopo mangiato.
Fonti e bibliografia
Si ringrazia il Dr. Tommaso Casotti, autore della prima stesura dell’articolo
- NIH
- NHS
- Systemic Sclerosis (Scleroderma) – Rotimi Adigun; Amandeep Goyal; Anis Hariz.
- Ghizzoni C, Sebastiani M, et al. Prevalence and evolution of scleroderma pattern at nailfold videocapillaroscopy in systemic sclerosis patients: Clinical and prognostic implications. Microvasc Res. 2015 May;99:92-5.
- Brady SM, Shapiro L, Mousa SA. Current and future direction in the management of scleroderma. Arch Dermatol Res. 2016 Sep;308(7):461-71.
Tutti gli aggiornamenti su salute, alimentazione e benessere.