Introduzione
La trisomia 13 è stata descritta per la prima volta come causa di sindrome clinica nel 1960, dal Dr. Patau; è una condizione caratterizzata dalla presenza di
- difetti cerebrali,
- assenza di uno o entrambi gli occhi (anoftalmia),
- difetti congeniti del viso (palatoschisi, labbro leporino),
- pollici a scatto,
- presenza di un numero di dita superiori alla norma (polidattilia)
- “voglia di fragola” (emangiomi capillari).
La sindrome di Patau viene oggi spesso diagnosticata già in fase prenatale, durante gli esami di screening, meno comunemente alla nascita; la causa è prettamente genetica ed il fattore di rischio più rilevante è un’età avanzata della mamma.
Per informazioni sui centri di diagnosi e cura, nonché sulle associazioni di pazienti sul territorio, è possibile rivolgersi al Telefono Verde Malattie Rare 800.89.69.49, attivo dal lunedì al venerdì dalle 9:00 alle 13:00.
Aspettativa di vita
Circa il 15% delle gravidanze in cui viene formulata una diagnosi di trisomia 13 si osserva un aborto spontaneo prima del parto.
Numerosi studi di grandi dimensioni hanno purtroppo dimostrato una prognosi sfavorevole, con una sopravvivenza mediana di 7-10 giorni nei pazienti nati vivi e, nel complesso, una sopravvivenza inferiore all’anno per il 90% dei neonati affetti (salvo recenti casi di sopravvivenza più lunga, come conseguenza di una terapia medica particolarmente aggressiva).
La ragione della scarsa aspettativa di vita risiede in genere nelle malformazioni cerebrali e cardiovascolari presenti.
Cause
La sindrome di Patau è una malattia genetica, ovvero causata dalla presenza di difetti sul DNA del neonato; si verifica in circa un neonato su 5000 (contro, ad esempio, 1/700 della sindrome di Down).
La sindrome di Patau si verifica per caso e non è causata da qualcosa che i genitori possono avere o non aver fatto. La maggior parte dei casi della sindrome non è familiare (non è ereditaria) ed è invece conseguenza di eventi imprevedibili al momento della produzione di spermatozoo od ovulo. Quello che si verifica è un errore durante la divisione cellulare dei gameti, che influisce gravemente sullo sviluppo del bambino nell’utero.
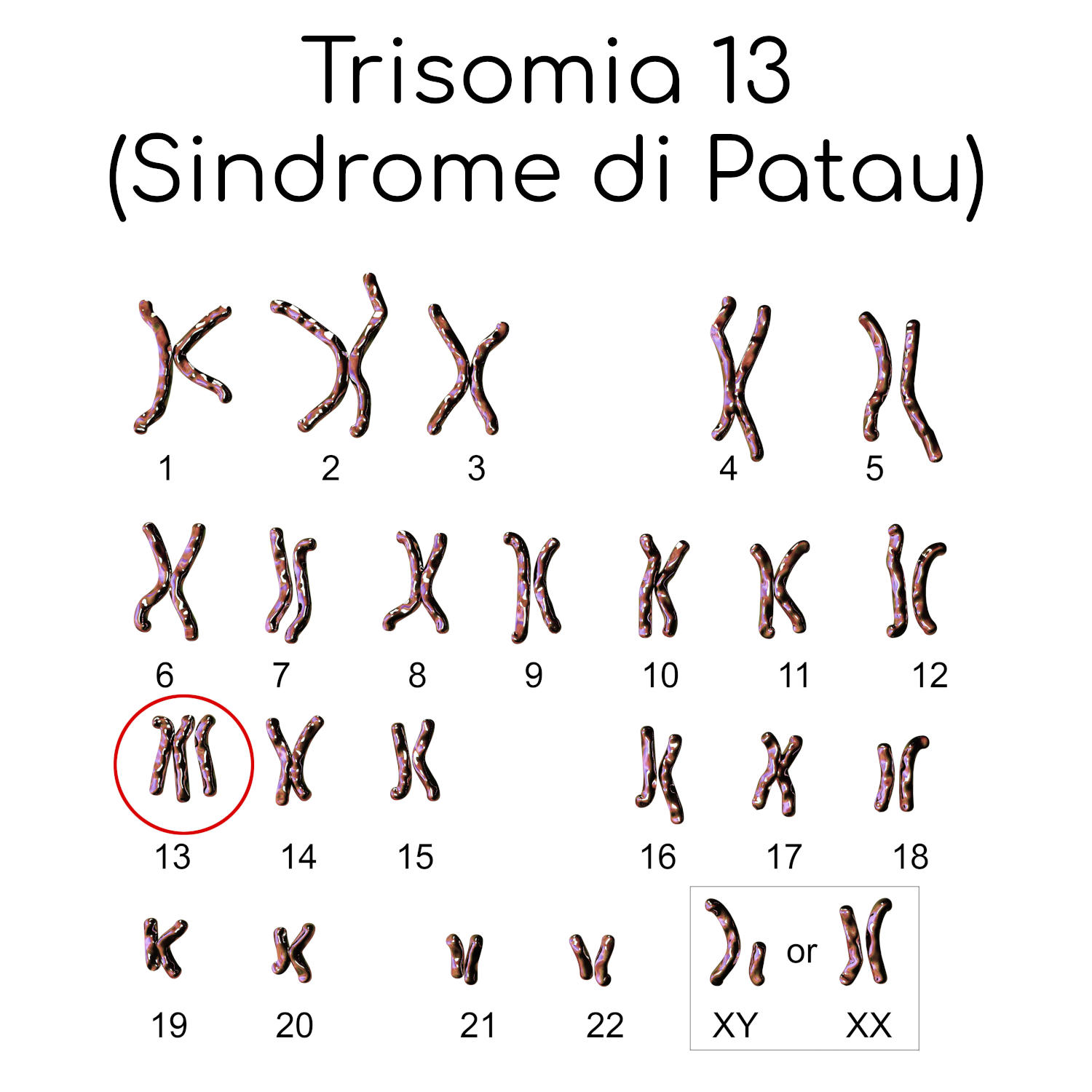
Più in particolare quello che si osserva è in genere la presenza anomala di tre copie del cromosoma 13 (da cui il nome trisomia 13), anziché le normali due (a causa della mancata separazione che dovrebbe avvenire durante la meiosi), sebbene siano stati descritti anche altri possibili difetti.
Il più importante fattore di rischio è un’età materna avanzata (superiore a 35 anni).
Shutterstock/Kateryna Kon
In circa il 10% dei casi il materiale genetico viene invece riorganizzato tra il cromosoma 13 e un altro cromosoma (traslocazione cromosomica).
In un ulteriore caso su 20 sono solo alcune cellule che hanno una copia in eccesso del cromosoma 13 (mosaicismo), oppure solo una parte di un cromosoma 13 è in eccesso (trisomia parziale 13).
I sintomi e i difetti dei neonati affetti da queste due anomalie genetiche sono generalmente meno gravi rispetto alla trisomia 13 tradizionale, con una sopravvivenza aumentata.
Sintomi e quadro clinico
I neonati con sindrome di Patau mostrano tipicamente un rallentamento della crescita intrauterina e una testa di dimensioni ridotte (microcefalia).
I difetti facciali comprendono:
- difetti dell’anatomia degli occhi:
- assenza completa di uno o entrambi i bulbi oculari in presenza di palpebre,congiuntiva e apparato lacrimale (anoftalmia),
- malformazione congenita dell’occhio, che risulta più piccolo rispetto alla norma (microftalmia),
- presenza di un’unica cavità orbitaria (che dovrebbe accogliere l’occhio) più o meno completa, localizzata in mezzo alla fronte (ciclopia),
- labbro leporino,
- palatoschisi,
- fronte inclinata,
- orecchie piccole e malformate,
- sordità congenita,
- mandibola di dimensioni e volume ridotti rispetto alla mascella (micrognazia).
Il cervello risulta malformato (oloprosencefalia alobare, ovvero assenza della normale separazione tra i due emisferi cerebrali) così come il cuore, che può essere affetto da:
- difetto del setto ventricolare/atriale e atrioventricolare (DIV e DIA, alterazioni delle pareti che separano le camere del cuore) e ,
- tetralogia di Fallot,
- ventricolo destro a doppia uscita.
Vale la pena notare che i soli difetti cardiaci in genere non sarebbero fatali durante, anche se non trattati.
I difetti tipici delle estremità comprendono
- un numero aumentato di dita,
- piede torto congenito (Piede equino-varo-supinato) o altra deformazione.
Si osservano infine anomalie d’organo che possono interessare polmoni, fegato, reni, tratto genito-urinario, tratto digerente e pancreas, tra i più comuni quelli genitali, ad esempio
- criptorchidismo,
- ipospadia,
- ipoplasia delle piccole labbra
- e utero bicorne.
I pochi pazienti sopravvissuti all’infanzia mostrano in genere gravi disturbi psicomotori, ritardo della crescita, disabilità intellettiva e convulsioni.
Diagnosi
La diagnosi della sindrome di Patau può essere fatta già in fase prenatale (prima del parto) mediante il prelievo dei villi coriali, l’amniocentesi o l’analisi del DNA fetale libero.
A meno di specifici fattori di rischio (come un’età superiore ai 35 anni) in genere si procede ad uno screening mediante esami non invasivi (ad esempio il test combinato, comprensivo di esame del sangue della mamma ed ecografia) e solo in caso di risultato preoccupante fa seguito la verifica con gli esami più invasivi (ma caratterizzati da una sensibilità decisamente maggiore).
Anche le normali ecografie prenatali (soprattutto quelle successive alla 17esima settimana) possono contribuire alla scoperta della malattia, consentendo di rilevare alcune delle anomalie tipiche del sistema nervoso, facciali, scheletriche, renali o cardiache, nonché il ritardo di crescita tipicamente manifestato dai feti affetti da trisomia 13.
Cura
Il trattamento intensivo della sindrome di Patau è controverso a causa della pessima prognosi dei neonati, a prescindere dalle terapie, considerabile quindi come forma di accanimento terapeutico.
Si opta più spesso per un trattamento volto a ridurre al minimo il dolore e le difficoltà incontrate dal bambino; al momento del parto potrebbe ad esempio aver bisogno di ossigenazione e ventilazione mediante approcci invasivi come l’intubazione o la tracheostomia, a causa di difetti facciali. I pazienti con difetti cardiaci possono richiedere un intervento chirurgico, così come potrebbe essere necessario il posizionamento di un tubo di alimentazione od interventi ortopedici correttivi.
In seguito sono spesso necessari piani di alimentazione specifica, prevenzione delle crisi epilettiche, profilassi antibiotica per le infezioni del tratto urinario e l’uso di apparecchi acustici.
Fonti e bibliografia
Tutti gli aggiornamenti su salute, alimentazione e benessere.






