Introduzione
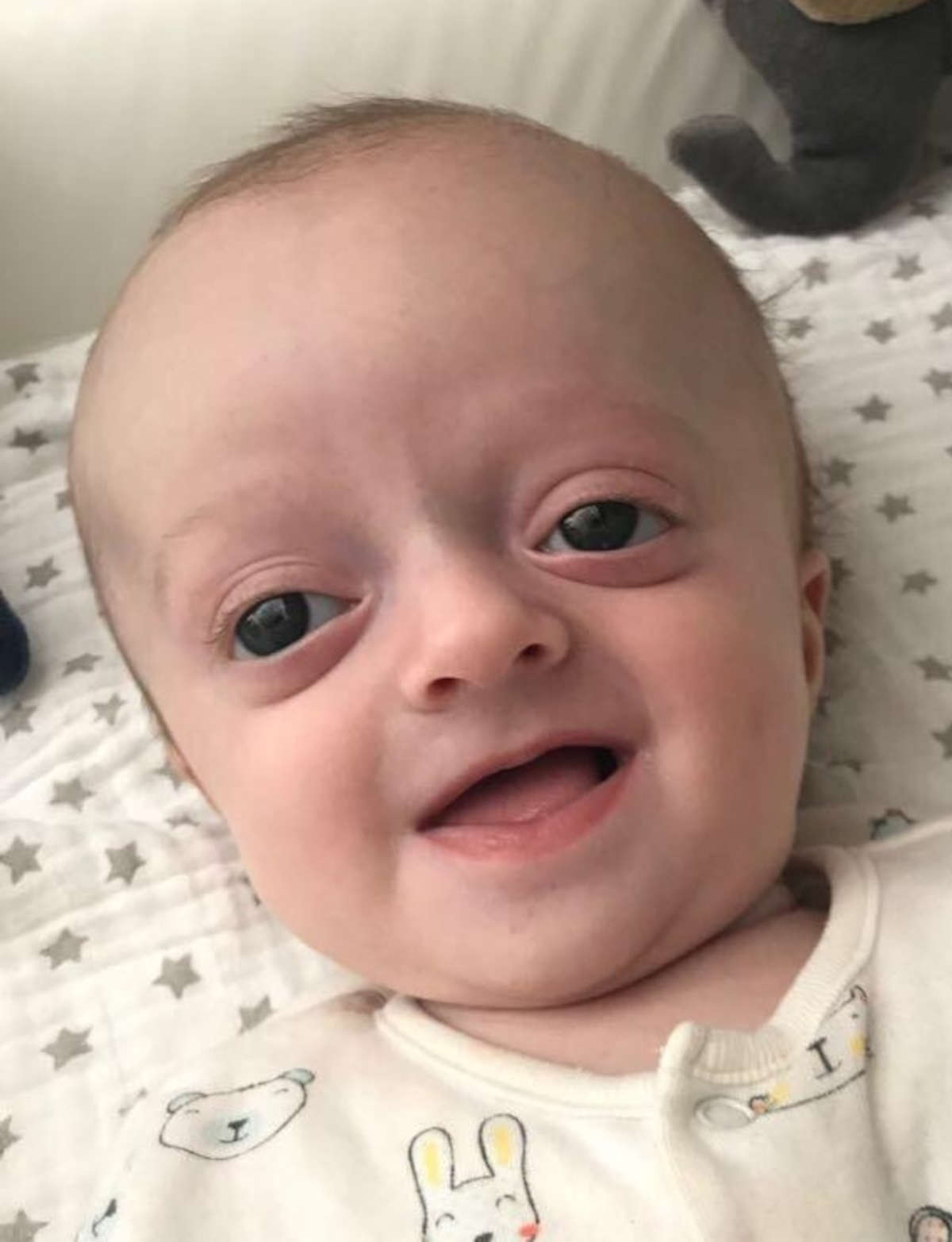
La sindrome di Crouzon è una malattia genetica caratterizzata dalla fusione prematura di alcune ossa del cranio (craniosinostosi). Questa fusione precoce impedisce al cranio di crescere normalmente, alterando la forma finale della testa e del viso, che causa ad esempio
- fronte prominente,
- occhi spalancati e sporgenti (e problemi di vista causati da orbite poco profonde, oltre che strabismo),
- naso curvo,
- mascella sottosviluppata (ipoplasia mascellare).
I pazienti affetti dalla sindrome di Crouzon possono sviluppare problemi dentali e perdita dell’udito (a causa di canali uditivi troppo stretti); in alcuni casi possono essere presenti labbro leporino e palatoschisi.
La gravità del quadro complessivo è variabile tra i diversi pazienti, ma tipicamente non si rileva alcun ritardo mentale.
La prognosi dei pazienti con sindrome di Crouzon dipende essenzialmente dalla tempestività di diagnosi e trattamento (quando necessario), in questo caso la maggior parte dei pazienti hanno un’aspettativa di vita sostanzialmente normale.
Immagini e fotografie

By Octave Crouzon – Octave Crouzon: Dysostose cranio-faciale héréditaire. Bulletins et mémoires de la Société médicale des hôpitaux de Paris Vol. 33, 545-55 (1912) (http://web2.bium.univ-paris5.fr/livanc/?cote=epo0192&do=chapitre), Public Domain, https://commons.wikimedia.org/w/index.php?curid=2049838

By Arthur J. Bedell, M.D. – “Oxycephalus”, The Journal of The American Medical Association, Vol. LXVIII, No. 26, Public Domain, https://commons.wikimedia.org/w/index.php?curid=130798871
Cause
La sindrome di Crouzon è una malattia genetica rara, che si osserva in circa 16 casi per milione di neonati (1 ogni 60000 neonati circa).
La mutazione genetica responsabile della malattia si trova sul cromosoma 10 ed è a carico del gene FGFR2, depositario della sequenza necessaria a sintetizzare una proteina indicata come “recettore del fattore di crescita dei fibroblasti di tipo 2”. Tra le sue molteplici funzioni questa proteina segnala alle cellule immature di diventare cellule ossee durante lo sviluppo embrionale.
Si ritiene che le mutazioni determinino la produzione di una proteina anomala ed eccessivamente attiva, che causa la fusione prematura delle ossa del cranio.
La sindrome di Crouzon viene trasmessa secondo il modello autosomico dominante, è cioè sufficiente un’unica copia alterata del gene (ereditata cioè da un unico genitore) per causare gli effetti della malattia. Questo significa che un genitore può trasmettere la mutazione ai propri figli con una probabilità del 50% (ogni figlio ha una probabilità del 50% di riceverla).
La sindrome ha penetranza completa, si manifesta cioè in tutti i neonati portatori della mutazione, ma espressività variabile, ovvero manifestandosi in modo più o meno visibile in pazienti diversi anche all’interno della stessa famiglia (dipendendo ad esempio da quali suture craniche sono coinvolte dal difetto, nonché dall’ordine e dalla velocità di progressione).
Si noti in ogni caso che in circa il 50% dei pazienti la mutazione è il risultato di mutazioni de novo, ovvero nate casualmente durante il concepimento e NON ereditate dai genitori.
Sintomi
La sindrome di Crouzon viene solitamente sospettata fin dalla nascita a causa di specifiche deformità facciali e craniche (oltre che, eventualmente, di un’anamnesi familiare positiva).
Le più comuni caratteristiche comprendono:
- appiattimento posteriore del cranio (brachicefalia), che assume un aspetto alto e stretto,
- aumento anomalo della distanza tra gli occhi (ipertelorismo oculare),
- occhi sporgenti (proptosi), che possono quindi non essere completamente protetti dalle palpebre e vengono perciò esposti al rischio di lesioni della congiuntiva e della cornea,
- fronte appiattita,
- naso a becco e mascella superiore sottosviluppata (con possibile malocclusione).
Questi pazienti possono anche soffrire di labbro leporino e/o palatoschisi, perdita dell’udito, strabismo e problemi dentali.
Mani e piedi appaiono del tutto normali e questo consente la diagnosi differenziale con altre craniosinostosi:
- sindrome di Apert, che condivide numerosi sintomi ma presenta alterazioni delle estremità (sindattilia)
- e la sindrome di Pfeiffer, che si manifesta anche con alluci/pollici corti e larghi.
Il decorso clinico del peggioramento delle deformità facciali nei primi 1-2 anni di vita aiuta a formulare la diagnosi clinica.
Diagnosi
La diagnosi è in genere clinica (osservando i sintomi), formulata fin dalla nascita e sostenuta dalla familiarità per la malattia. Nei casi dubbi è possibile ricorrere la verifica mediante esami genetici.
Cura
La gestione qualsiasi craniosinostosi, termine con cui si indicano difetti congeniti in cui una o più delle suture craniche si chiudono prematuramente durante lo sviluppo, è complessa e richiede un team multidisciplinare di specialisti che comprende tra l’altro pediatri, chirurghi maxillofacciali, chirurghi plastici, neurochirurghi, otorinolaringoiatri e oculisti.
La gestione chirurgica è normalmente indispensabile per la correzione dei difetti anatomici che possono causare cecità e disabilità intellettiva rispettivamente per impedimenti legati allo sviluppo orbitale (le orbite accolgono gli occhi nel cranio) e del cervello.
Se le anomalie della volta cranica vengono corrette tempestivamente i pazienti con sindrome di Crouzon potranno sviluppare una normale funzione cognitiva oltre che prevenire disturbi di vista e udito.
Avranno inoltre un’aspettativa di vita del tutto normale.
L’intervento ha l’obiettivo di garantire che vi sia spazio sufficiente all’interno del cranio per la crescita del cervello in via di sviluppo, ma non si rivela necessario per tutti i pazienti, viene cioè praticato solo quando il rischio di sviluppare complicanze legate alla sindrome giustifichi i rischi chirurgici; generalmente, se necessario, viene programmato tra i 4 e i 12 mesi di vita, prima in caso di ipertensione endocranica.
La condizione richiede comunque un monitoraggio a lungo termine soprattutto per quanto riguarda lo sviluppo oculare, a causa della propensione a sviluppare l’ambliopia strabica.
A causa delle anomalie delle vie aeree anche l’apnea notturna viene attentamente valutata nel tempo (mediante polisonnografia).
Circa il 30% dei pazienti con sindrome di Crouzon sviluppa idrocefalo, una pericolosa condizione caratterizzata da alterazioni del flusso del liquido cerebrospinale che circola attraverso le cavità (ventricoli) del cervello e del canale spinale, all’interno del cranio; l’aumento della pressione del fluido all’interno del cranio richiede un rapido intervento chirurgico.
Fonti e bibliografia
- MedLinePlus
- Crouzon Syndrome – Christopher D. Conrady; Bhupendra C. Patel
- RareDiseases
- Ospedale Bambino Gesù
Tutti gli aggiornamenti su salute, alimentazione e benessere.






