Introduzione
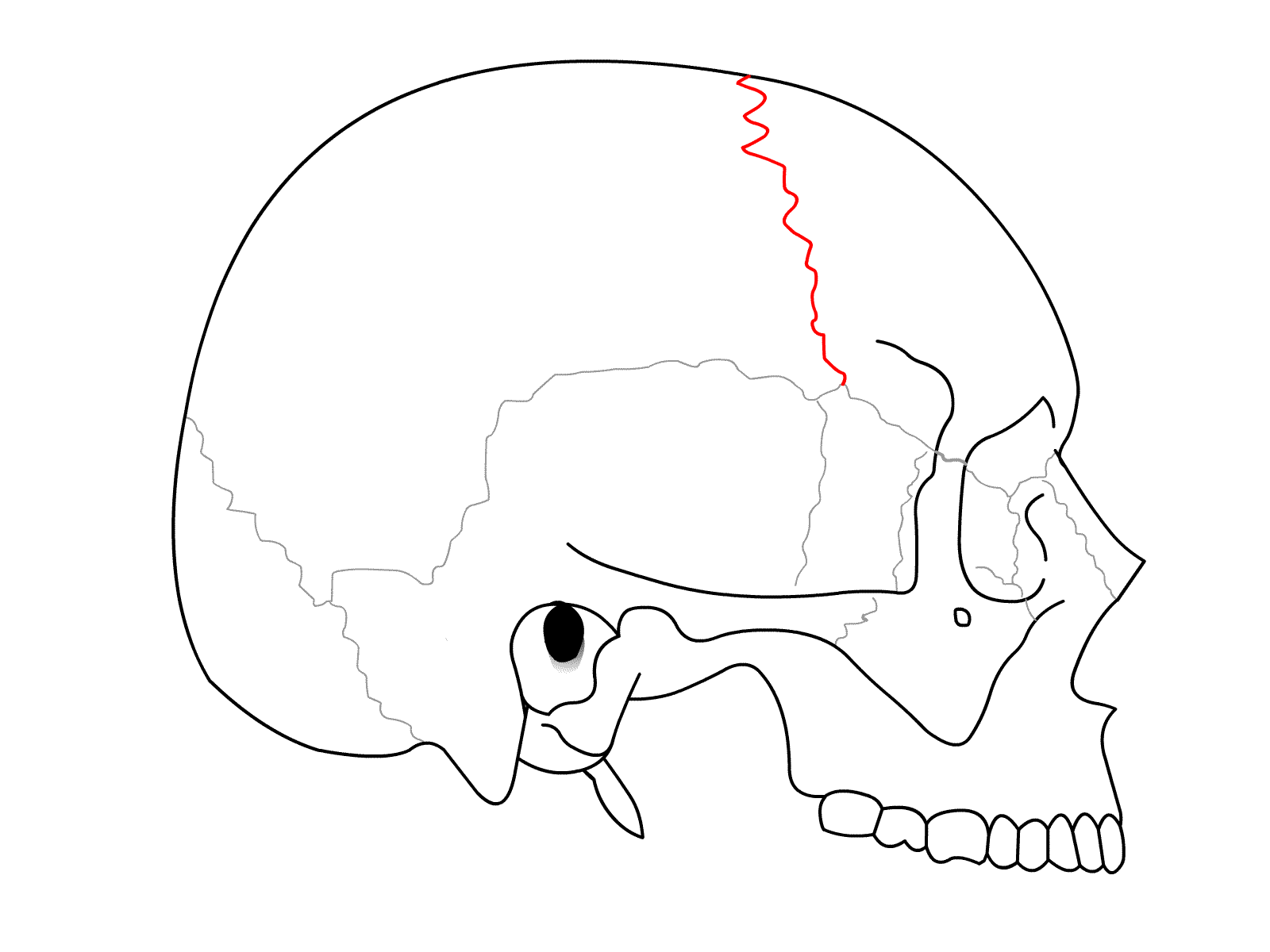
Parlando di cranio, ovvero delle ossa che racchiudono e proteggono il cervello, le suture sono le bande di tessuto che uniscono tra loro le diverse ossa di cui è composto; rappresentano il mezzo con cui il cranio può gradualmente espandersi con il progressivo aumento del volume del cervello, tanto durante la gravidanza quanto durante la crescita del bambino, indurendosi infine a maturazione terminata. Dopo la chiusura delle suture il cranio non è quindi più in grado di espandersi ulteriormente.
La craniosinostosi è una condizione caratterizzata da una chiusura con eccessivo anticipo delle suture, che limita così la possibilità per il cervello di espandersi fino a raggiungere forma e dimensioni normali.
La sindrome di Apert ne è un triste esempio ed è una malattia genetica, che si manifesta con caratteristiche anomalie estetiche del viso, degli arti, problemi dentali, disturbi della vista e respiratori e spesso anche ritardo mentale.
Cause
La sindrome di Apert è una malattia rara, che si stima verificarsi con una frequenza compresa tra 1 su 65.000-200.000 nascite, a seconda dello studio.
Le mutazioni responsabili della sindrome di Apert si localizzano su un gene noto come FGFR2, che è responsabile delle istruzioni per la sintesi di una proteina chiamata “recettore 2 del fattore di crescita dei fibroblasti”. Tra le sue molteplici funzioni la proteina FGFR2 svolge un ruolo chiave nello sviluppo prima della nascita segnalando alle cellule immature di trasformarsi in cellule ossee.
La mutazione in una parte specifica del gene FGFR2 altera la proteina e, da un punto di vista pratico, amplifica il segnale inducendo ad una maturazione troppo rapida, che promuove in ultimo la fusione prematura delle ossa di cranio, mani e piedi.
Maschi e femmine sono ugualmente colpiti e l’incidenza della malattia aumenta significativamente con l’età paterna (si ritiene che fornisca un vantaggio selettivo agli spermatozoi portatori del difetto genetico).
La sindrome di Apert ha
- penetranza completa (tutti gli individui affetti dall’anomalia genetica mostrano i sintomi della malattia)
- ma espressività variabile (alcuni pazienti sviluppano manifestazioni molto lievi mentre altri soffrono di gravissime disabilità).
Da un punto di vista genetico la condizione è autosomica dominante:
- autosomica: il difetto genetico NON è relativo a uno dei due cromosomi sessuali,
- dominante: è sufficiente un unico gene mutato (ad esempio quello ricevuto dal padre).
Quasi tutti i casi derivano da nuove mutazioni, che si verificano cioè durante la formazione delle cellule riproduttive (ovuli o sperma) in uno dei due genitori, oppure durante le primissime fasi dello sviluppo embrionale.
Sintomi
La sindrome di Apert è una malattia genetica caratterizzata da anomalie scheletriche, in cui spicca una chiusura prematura delle ossa del cranio (craniosinostosi) che
- impedisce al cranio di crescere normalmente,
- influisce sulla forma della testa e del viso.
È proprio la craniosinostosi la causa della maggior parte delle tipiche caratteristiche facciali della sindrome, come ad esempio
- testa con forma anomala, più piccola del normale (più in particolare la larghezza prevale sulla lunghezza),
- fonte ampia,
- occhi sporgenti a causa di orbite non sufficientemente profonde e distanti tra loro, con possibili disturbi della vista (tra cui strabismo),
- anomalie del naso (radice nasale depressa, deviazione del setto),
- ridotto sviluppo della mascella (con difetti della dentatura e palatoschisi).
Le capacità cognitive nelle persone con sindrome di Apert vanno dal normale ad una disabilità intellettiva lieve o moderata.
L’altra caratteristica tipica della condizione è la sindattilia, ovvero la presenza di dita (di mani e/o piedi) fuse insieme; la gravità della fusione è variabile, sebbene le mani tendano ad essere più gravemente colpite rispetto ai piedi. Più raramente si osserva polidattilia, ovvero la presenza di un numero di dita superiore a 10.
Alcuni pazienti sviluppano anomalie ossee a livello di gomiti o spalle, che possono limitare i movimenti e impedire l’esecuzione delle normali attività quotidiane; i difetti possono presentarsi su un solo lato del corpo o su entrambi.
A queste anomalie si associano spesso altri disturbi, come ad esempio
- disturbi di udito e infezioni ricorrenti (otiti) dovute a malformazioni della struttura dell’orecchio,
- blocco parziale delle vie aeree e portare a difficoltà respiratorie a causa dello sviluppo anomalo del viso e della testa,
- sudorazione insolitamente abbondante (iperidrosi),
- pelle grassa con acne grave,
- zone di peli mancanti nelle sopracciglia.
Complicazioni
Le principali complicanze che possono verificarsi nei pazienti con sindrome di Apert comprendono:
- Aumento della pressione intracranica, che può causare papilledema (lesione del nervo ottico) e deterioramento cognitivo
- Disturbi visivi
- Complicanze respiratorie
- Lesione del midollo spinale e deficit neurologici in pazienti con anomalie della colonna vertrebrale (ad esempio fusione delle vertebre)
- Polmonite ab ingestis ed altre complicazioni polmonari
Diagnosi
La diagnosi della sindrome di Apert è clinica, basata cioè sulle caratteristiche fisiche peculiari.
Nei casi dubbi, ad esempio in assenza di altri casi familiari di malattia, è possibile ricorrere ad esami di imaging (TAC e risonanza magnetica) che possano confermare la presenza di craniosinostosi o altre anomalie scheletriche; questi stessi esami possono essere utili anche per rilevare complicazioni legate alla sindrome, come l’aumento della pressione intracranica.
Per dirimere ogni dubbio è in molti casi possibile ricorrere a test genetici.
Diagnosi prenatale
I test genetici prenatale, la risonanza magnetica e l’ecografia possono talvolta consentire la diagnosi già durante la gravidanza; il ricorso all’amniocentesi e/o alla villocentesi può essere utile, ma in genere non necessario salvo i casi più dubbi.
Cura
Come per altre malattie caratterizzate da craniosinostosi, la gestione del paziente è fortemente multidisciplinare, comprendendo tra l’altro
- pediatri,
- neurochirurghi,
- chirurghi plastici,
- chirurghi craniofacciali,
- oftalmologi
- e dentisti.
By I, RosarioVanTulpe, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=2421006
La chirurgia è necessaria per prevenire la chiusura completa della sutura coronale e proteggere così lo sviluppo cerebrale e si procede in genere prima dell’anno di età, seppure non esistano linee guida basate su dati ad indicare il momento ideale; allo stesso modo non esiste uno standard di cura per il trattamento della sindattilia (fusione delle dita), ma in genere si procede a ripetute correzioni chirurgiche con la crescita.
Il monitoraggio a lungo termine è essenziale per ridurre il rischio di sviluppare complicazioni legate alle craniosinostosi come strabismo, apnea notturna e pressione intracranica elevata, anche se sfortunatamente non è possibile prevenirne sempre l’insorgenza.
Sono allo studio farmaci in grado di supplire durante lo sviluppo alle anomalie genetiche responsabili della sindrome, ma purtroppo ancora non disponibili per l’uso sull’uomo.
Fonti e bibliografia
- Apert Syndrome – Christopher D. Conrady; Bhupendra C. Patel; Sandeep Sharma
- MedLinePlus
Tutti gli aggiornamenti su salute, alimentazione e benessere.






