Introduzione
La sferocitosi ereditaria è una malattia caratterizzata da anemia emolitica, una condizione che si verifica quando i globuli rossi vengono distrutti prima del previsto.
È causata da alterazioni genetiche, diverse a seconda della porzione di DNA interessata, che compromettono alcune proteine della membrana dei globuli rossi. Le cellule perdono progressivamente la normale forma biconcava e diventano più sferiche, rigide e fragili.
È per definizione presente sin dalla nascita e l’insieme dei sintomi può variare da quadri lievi a molto gravi, comprendendo ad esempio
- pallore,
- affaticamento e stanchezza,
- ittero (pelle e parte bianca degli occhi di colore giallasto),
- formazione di calcoli biliari,
- ingrossamento della milza (splenomegalia).
Nei casi più severi si possono verificare inoltre
- disturbi dello sviluppo
- bassa statura,
- pubertà ritardata,
- e, più raramente, anomalie scheletriche.
Il trattamento dipende dalla gravità. Le forme lievi possono richiedere soltanto controlli periodici, mentre negli altri casi possono essere indicati l’integrazione di folati, le trasfusioni di sangue e, in pazienti selezionati, la rimozione chirurgica totale o parziale della milza. La splenectomia riduce l’emolisi, ma comporta rischi permanenti che devono essere attentamente valutati.
La prognosi generale è considerata buona, anche se variabile a seconda della gravità della malattia.
Cause
I globuli rossi normali possiedono una caratteristica forma biconcava, che rappresenta un compromesso ideale in termini di rapporto superficie-volume e capacità di adattamento all’ambiente in cui si trovano a operare.
La sferocitosi ereditaria è il risultato di una carenza o di un’anomalia di una o più proteine necessarie al mantenimento di un’adeguata adesione tra il citoscheletro, responsabile della struttura interna della cellula, e la membrana. Poiché la superficie cellulare non è più adeguatamente sostenuta dal citoscheletro, la cellula perde parte della membrana e assume una forma sferica anormale, con una riduzione del rapporto superficie-volume. Diventa così meno deformabile e più vulnerabile alla distruzione.
Lo stesso termine sferocitosi indica questa anomalia di forma, che passa dall’essere quella di un disco biconcavo, simile a una ciambella, a una sfera. I globuli rossi così prodotti sono detti sferociti. La loro scarsa deformabilità ne favorisce il trattenimento e la distruzione nella milza, causando i sintomi tipici dell’anemia emolitica.
La ragione ultima di questi difetti è da ricercarsi in alterazioni genetiche delle porzioni di DNA contenenti le istruzioni per la sintesi delle proteine di struttura.
La sferocitosi ereditaria è la malattia emolitica genetica più comune, soprattutto nelle popolazioni di origine nord-europea, con una diffusione stimata di circa 1 persona su 2.000.
Trasmissione genetica
La sferocitosi ereditaria è una malattia genetica, ovvero causata dall’alterazione di uno dei geni coinvolti nella struttura della membrana dei globuli rossi. I geni fanno parte del DNA, il materiale genetico di base che si trova nelle cellule del nostro corpo. Ogni gene svolge una funzione diversa e alcuni contengono le istruzioni necessarie per produrre proteine.
I principali geni coinvolti in questa condizione sono:
- SLC4A1,
- SPTA1,
- SPTB,
- ANK1,
- EPB42.
Ogni essere umano eredita due copie della maggior parte dei geni, una da ciascun genitore.
In circa il 75% dei casi la sferocitosi ereditaria si trasmette con modalità autosomica dominante. I casi rimanenti comprendono nuove alterazioni genetiche, dette de novo, e forme autosomiche recessive, meno comuni. Il termine autosomico indica che i geni interessati non si trovano sui cromosomi sessuali, mentre la differenza tra modello dominante e recessivo può essere così spiegata:
- Dominante significa che è sufficiente una sola copia alterata del gene responsabile per manifestare la malattia. Ciascun figlio di un genitore che possiede la variante ha quindi una probabilità del 50%, ossia 1 su 2, di ereditarla. La gravità può comunque variare anche tra membri della stessa famiglia.
- Recessiva significa che, per manifestare la malattia, è necessario che entrambe le copie dello stesso gene siano alterate. Quando due portatori della stessa malattia autosomica recessiva hanno figli, per ogni gravidanza esiste una probabilità del 25% di concepire un figlio affetto, del 50% di concepire un figlio portatore e del 25% di concepire un figlio che non abbia ereditato nessuna delle due varianti.
L’alterazione genetica può essere ereditata da un genitore oppure comparire per la prima volta nella persona affetta. Una consulenza genetica può aiutare la famiglia a comprendere la specifica modalità di trasmissione e il rischio per eventuali figli.
Sintomi
La malattia può manifestarsi fin dal periodo neonatale, spesso con ittero, oppure essere riconosciuta durante l’infanzia o soltanto in età adulta nelle forme più lievi. Si rileva una grande variabilità tra pazienti differenti e la malattia viene in genere distinta in base alla gravità in:
- lieve, spesso diagnosticata più tardi,
- moderata,
- grave.
La triade classica di sintomi della sferocitosi è costituita da
- ittero, ossia colorazione gialla di pelle e sclere degli occhi,
- anemia
- stanchezza,
- pallore,
- ridotta tolleranza allo sforzo,
- ingrandimento della milza
- dolore addominale o senso di peso nella parte superiore sinistra dell’addome.
È tuttavia raro osservare tutti questi segni nei neonati, nei quali il reperto più comune è l’ittero. Quest’ultimo può avere anche cause del tutto fisiologiche nei primi giorni di vita, come spiegato nell’articolo dedicato.
Complicazioni
La colelitiasi, ossia la formazione di calcoli biliari nella cistifellea, è una delle complicanze più comuni. Può comparire già durante l’infanzia o l’adolescenza, anche se la frequenza tende ad aumentare con l’età.
Alcuni pazienti possono anche presentare crisi ematologiche, che possono essere
- emolitiche, con un improvviso aumento della distruzione dei globuli rossi, spesso in concomitanza con un’infezione,
- aplastiche, con una rapida caduta dell’emoglobina dovuta alla temporanea riduzione della produzione dei globuli rossi da parte del midollo osseo; la causa tipica è un’infezione da parvovirus B19,
- megaloblastiche, dovute a una marcata carenza di folati e oggi poco comuni quando l’apporto è adeguato.
Alcuni pazienti, soprattutto nelle forme più gravi e non adeguatamente controllate, possono manifestare disturbi della crescita.
Diagnosi
La diagnosi di sferocitosi ereditaria viene formulata integrando storia clinica, visita medica ed esami del sangue. Non esiste un singolo test in grado di riconoscere con certezza tutte le forme della malattia, perciò i risultati devono essere interpretati nel loro insieme, preferibilmente da un ematologo o da un centro con esperienza nelle anemie congenite.
Valutazione clinica
Il medico raccoglie informazioni su ittero neonatale, episodi di anemia, stanchezza, calcoli biliari, crisi associate a infezioni e precedenti trasfusioni. È importante ricostruire anche la storia familiare, cercando casi di anemia, ittero ricorrente, calcoli biliari in giovane età o interventi di rimozione della milza.
Durante la visita vengono valutati il pallore, l’ittero e l’eventuale ingrossamento della milza. Nei bambini si controllano inoltre crescita e sviluppo. L’assenza di una storia familiare non esclude la malattia, perché possono verificarsi alterazioni genetiche nuove o forme recessive.
Esami del sangue
Gli accertamenti iniziali comprendono normalmente:
- emocromo completo con indici dei globuli rossi;
- conteggio dei reticolociti, globuli rossi giovani che aumentano quando il midollo cerca di compensare l’emolisi;
- striscio di sangue periferico, osservato al microscopio;
- bilirubina, in particolare la frazione indiretta;
- lattato deidrogenasi o LDH e aptoglobina, utilizzate insieme agli altri parametri per documentare l’emolisi;
- test diretto dell’antiglobulina, noto anche come test di Coombs diretto, che nella sferocitosi ereditaria è generalmente negativo e aiuta a distinguere la malattia da alcune anemie emolitiche autoimmuni.
L’emoglobina può essere normale nelle forme lievi e ridotta in quelle più importanti. I reticolociti sono in genere aumentati, mentre la bilirubina indiretta può essere elevata e l’aptoglobina ridotta. Il volume corpuscolare medio, o MCV, può essere normale o lievemente ridotto, ma non è un criterio diagnostico affidabile se considerato da solo. La concentrazione media di emoglobina, o MCHC, è spesso aumentata, ma può risultare normale e può essere influenzata anche da problemi tecnici del campione.
Lo striscio può mostrare sferociti, globuli rossi rotondi e privi della tipica area chiara centrale. La loro presenza sostiene il sospetto, ma non è esclusiva della sferocitosi ereditaria e può essere poco evidente nelle forme lievi o nei neonati.
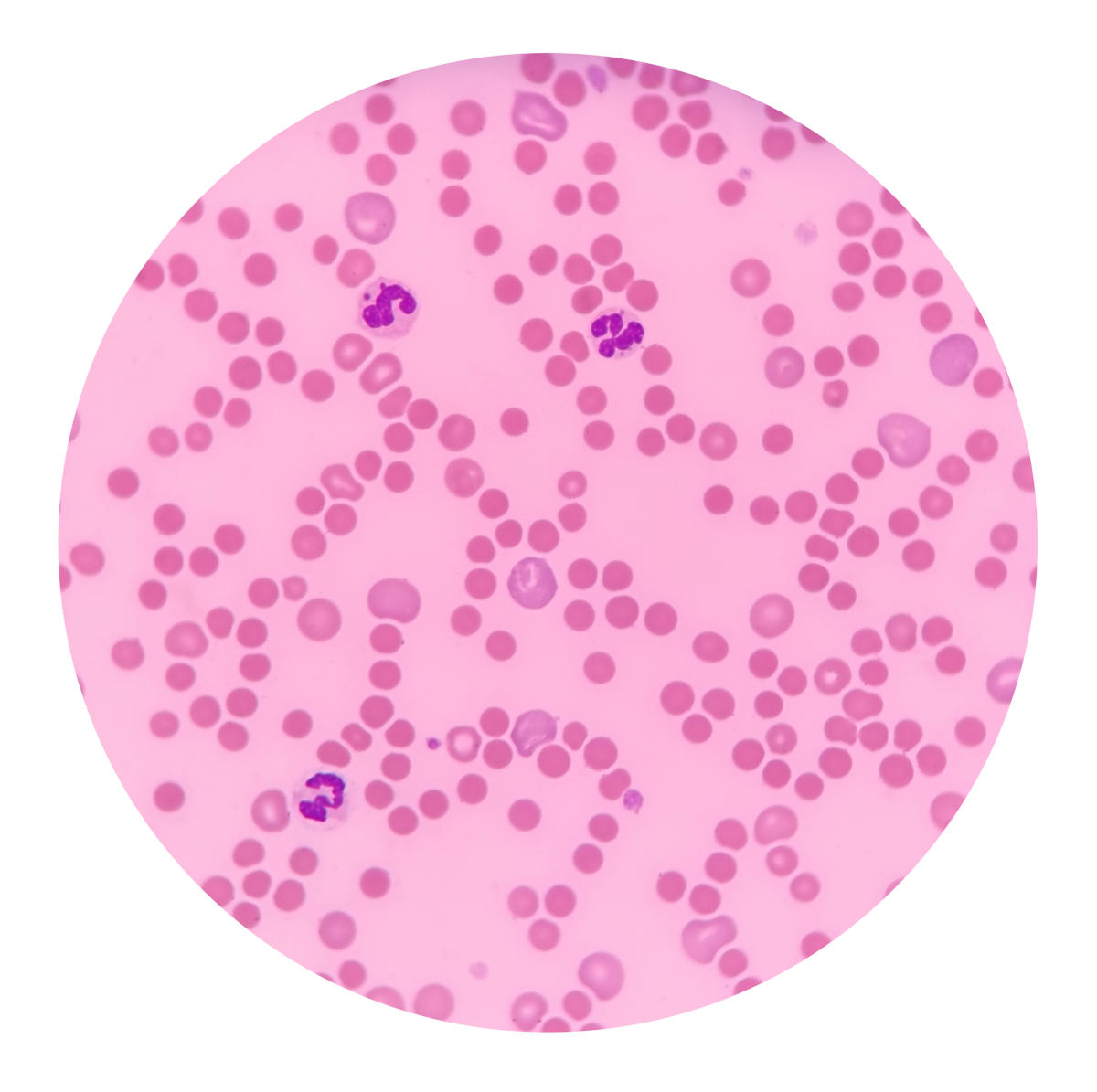
Shutterstock/Pee Paew
Test di conferma
Quando il quadro clinico e gli esami di base non sono sufficienti, si ricorre a test specifici sulla membrana dei globuli rossi. Uno dei più utilizzati è il test di legame con eosina-5-maleimide, o test EMA, eseguito mediante citometria a flusso. Nella sferocitosi la fluorescenza risulta in genere ridotta.
Il test EMA è accurato, ma non infallibile: può dare risultati normali in alcuni pazienti e alterati in altre rare malattie della membrana. In base alla disponibilità del laboratorio può essere associato a prove di fragilità osmotica, come il test di lisi in glicerolo. Le prove tradizionali eseguite con soluzioni saline hanno prestazioni inferiori e non dovrebbero essere utilizzate come unico criterio diagnostico.
L’ektacitometria, che misura la deformabilità dei globuli rossi in diverse condizioni osmotiche, può aiutare a distinguere la sferocitosi da altre malattie della membrana, ma è disponibile soltanto in centri specializzati. L’analisi biochimica delle proteine di membrana viene utilizzata raramente.
Una trasfusione recente può interferire con alcuni di questi test perché nel sangue circolano anche globuli rossi del donatore. È quindi importante informare il laboratorio e programmare gli accertamenti con lo specialista.
Analisi genetica
L’analisi genetica non è necessaria in tutti i pazienti con un quadro clinico e di laboratorio tipico. Può essere indicata quando la diagnosi resta incerta, la presentazione è particolarmente grave o atipica, gli esami specifici forniscono risultati discordanti oppure è importante distinguere la sferocitosi da altre anemie ereditarie prima di prendere decisioni terapeutiche, soprattutto prima di una splenectomia.
Il test genetico può inoltre essere utile per la consulenza familiare, ma un risultato negativo non esclude sempre la malattia, perché non tutte le alterazioni sono facilmente identificabili o interpretabili.
Diagnosi differenziale e altri accertamenti
Gli sferociti possono comparire anche nell’anemia emolitica autoimmune e, nei neonati, in alcune incompatibilità tra il sangue materno e quello del bambino. Devono inoltre essere considerate altre malattie ereditarie della membrana, come le stomatocitosi, le carenze enzimatiche dei globuli rossi, le emoglobinopatie e alcune anemie congenite caratterizzate da una produzione inefficace delle cellule del sangue.
La distinzione è importante perché la splenectomia, utile in alcuni pazienti con sferocitosi, può essere inefficace o pericolosa in altre malattie della membrana. Nei casi dubbi è quindi indicata una valutazione ematologica specialistica.
L’ecografia addominale non conferma da sola la diagnosi. Può essere richiesta per misurare la milza o cercare calcoli nella cistifellea, soprattutto in presenza di dolore addominale, ittero più intenso o prima di un eventuale intervento. L’esame del midollo osseo non è normalmente necessario ed è riservato a situazioni selezionate in cui si sospettano altre patologie.
Cura
Gli obiettivi del trattamento sono mantenere un livello adeguato di emoglobina, ridurre stanchezza, ittero e altri disturbi, sostenere la crescita nei bambini e prevenire o gestire complicanze come crisi anemiche e calcoli biliari. La scelta dipende dalla gravità, dall’età, dal fabbisogno di trasfusioni, dalla qualità di vita e dalle altre condizioni di salute.
Non esiste una terapia farmacologica capace di correggere il difetto genetico. Molte persone con forme lievi non hanno bisogno di un trattamento continuativo e possono condurre una vita normale con controlli periodici. La splenectomia riduce efficacemente la distruzione dei globuli rossi, ma non elimina il difetto della loro membrana e non è indicata per tutti.
Osservazione e folati
Nelle forme lievi è generalmente sufficiente la sorveglianza clinica. L’acido folico può essere prescritto nelle forme moderate o gravi, durante periodi di maggiore produzione dei globuli rossi e in circostanze particolari, come la gravidanza. Non è necessariamente utile in tutte le forme lievi e non dovrebbe essere assunto a lungo senza indicazione medica.
Gli integratori di ferro non trattano la sferocitosi e devono essere utilizzati soltanto se gli esami confermano una reale carenza. Un’assunzione non necessaria può favorire un accumulo di ferro, rischio che deve essere controllato soprattutto nelle persone sottoposte a numerose trasfusioni.
Trattamento dell’anemia e delle crisi
Le trasfusioni di globuli rossi vengono utilizzate in caso di anemia grave o sintomatica. Sono più frequentemente necessarie nei neonati e nei lattanti con forme importanti, durante una crisi aplastica o quando l’organismo non riesce a compensare un improvviso aumento dell’emolisi. La decisione non dipende soltanto dal valore dell’emoglobina, ma anche dai sintomi, dall’età e dalle condizioni generali.
Nei neonati l’ittero deve essere controllato attentamente. Se la bilirubina raggiunge livelli pericolosi possono essere necessarie la fototerapia e, nei casi più gravi, procedure trasfusionali specifiche per prevenire danni neurologici.
In alcuni lattanti selezionati lo specialista può valutare per un periodo limitato l’eritropoietina, un ormone che stimola la produzione dei globuli rossi, con l’obiettivo di ridurre il ricorso alle trasfusioni. Non è un trattamento di routine per bambini più grandi o adulti e richiede un attento monitoraggio ematologico.
Una febbre accompagnata da improvviso pallore, marcata debolezza, respiro affannoso, battito accelerato o sonnolenza può indicare una crisi aplastica o emolitica e richiede una rapida valutazione medica. Il parvovirus B19, causa frequente di crisi aplastica, può essere particolarmente rischioso anche per le persone in gravidanza che entrano in contatto con il paziente.
Splenectomia
La splenectomia consiste nella rimozione totale o parziale della milza, principale sede di distruzione degli sferociti. In genere aumenta l’emoglobina e riduce nettamente l’emolisi, l’ittero e il fabbisogno di trasfusioni. Gli sferociti rimangono tuttavia presenti perché l’intervento non corregge il difetto genetico.
L’intervento è normalmente raccomandato nelle forme gravi e dipendenti dalle trasfusioni. Nelle forme moderate viene valutato caso per caso, considerando crisi ripetute, ridotta tolleranza allo sforzo, crescita compromessa, splenomegalia dolorosa, riduzione delle altre cellule del sangue e impatto sulla qualità di vita. Nelle forme lievi non è generalmente indicato.
Nei bambini, quando possibile, l’intervento viene rinviato almeno oltre i 5-6 anni, perché il rischio di infezioni gravi dopo la rimozione della milza è maggiore nei più piccoli. La decisione deve essere condivisa tra famiglia, ematologo e chirurgo esperto.
La splenectomia totale offre in genere un miglior controllo dell’emolisi, ma espone per tutta la vita a un maggiore rischio di infezioni invasive, in particolare da pneumococco, meningococco ed Haemophilus influenzae di tipo b. Può inoltre aumentare il rischio di trombosi in alcuni pazienti. La splenectomia parziale conserva una parte della funzione immunitaria della milza, ma può correggere l’anemia in modo meno completo e il tessuto residuo può aumentare nuovamente di volume.
Prima di una splenectomia programmata devono essere completate, nei tempi stabiliti dal centro, le vaccinazioni raccomandate per le persone con asplenia. Dopo l’intervento possono essere necessari richiami vaccinali e una profilassi antibiotica, la cui durata varia in base all’età e al rischio individuale. La febbre dopo splenectomia deve essere considerata un’urgenza: è necessario contattare subito un medico, anche se le vaccinazioni sono aggiornate.
Calcoli biliari
I calcoli della cistifellea che causano coliche, infiammazione o altre complicanze vengono generalmente trattati con la colecistectomia, ossia la rimozione della cistifellea, di solito per via laparoscopica. I calcoli privi di sintomi non richiedono automaticamente un intervento: la decisione viene personalizzata in base all’età, alle caratteristiche dei calcoli e all’eventuale programmazione di una splenectomia.
Dolore intenso nella parte superiore destra dell’addome, febbre, vomito, urine scure o un improvviso aumento dell’ittero richiedono una valutazione medica tempestiva.
Stile di vita
Non è prevista una dieta specifica. È consigliabile seguire un’alimentazione equilibrata, mantenere una buona idratazione e non assumere ferro, folati o altri integratori senza averne discusso con il medico. In presenza di una milza molto ingrossata vanno evitati sport di contatto e attività con rischio di trauma addominale, perché potrebbero provocarne la rottura.
Le normali vaccinazioni devono essere mantenute aggiornate. Chi è stato sottoposto a splenectomia dovrebbe portare con sé una tessera o un documento che segnali l’assenza della milza e ricevere istruzioni precise su come comportarsi in caso di febbre, viaggi o morsi di animali.
Controlli nel tempo
La frequenza dei controlli viene stabilita in base alla gravità. Il follow-up può comprendere visita medica, emocromo, reticolociti, bilirubina e valutazione delle riserve di ferro. Nei bambini si controllano crescita e sviluppo; l’ecografia della cistifellea viene eseguita in presenza di sintomi o secondo il programma del centro curante.
È opportuno rivolgersi a un ematologo in caso di diagnosi incerta, anemia moderata o grave, trasfusioni ripetute, crisi ematologiche, crescita rallentata, calcoli biliari o valutazione di una splenectomia. Anche la gravidanza richiede un confronto con l’ematologo e il ginecologo per programmare controlli e integrazioni appropriate.
Fonti e bibliografia
- Hereditary Spherocytosis – Edgar A. Zamora; Catherine A. Schaefer
- RareDiseases
Tutti gli aggiornamenti su salute, alimentazione e benessere.






