Introduzione
L’atrofia muscolare spinale (acronimo inglese SMA, Spinal Muscolar Atrophy) è una patologia genetica ed ereditaria neuro-degenerativa che coinvolge esclusivamente i motoneuroni del midollo spinale, che vengono progressivamente a perdersi.
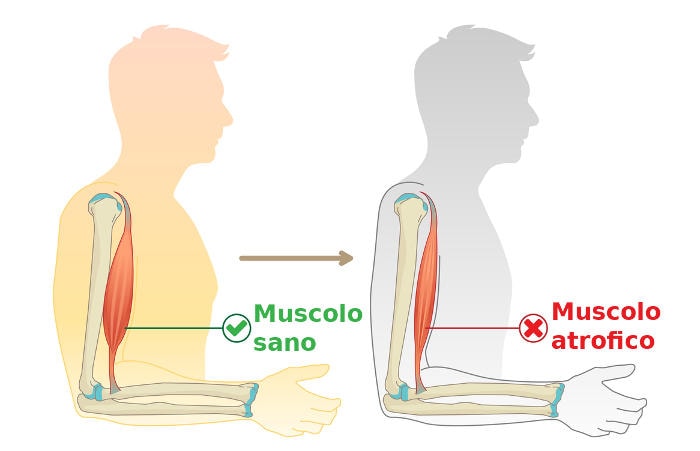
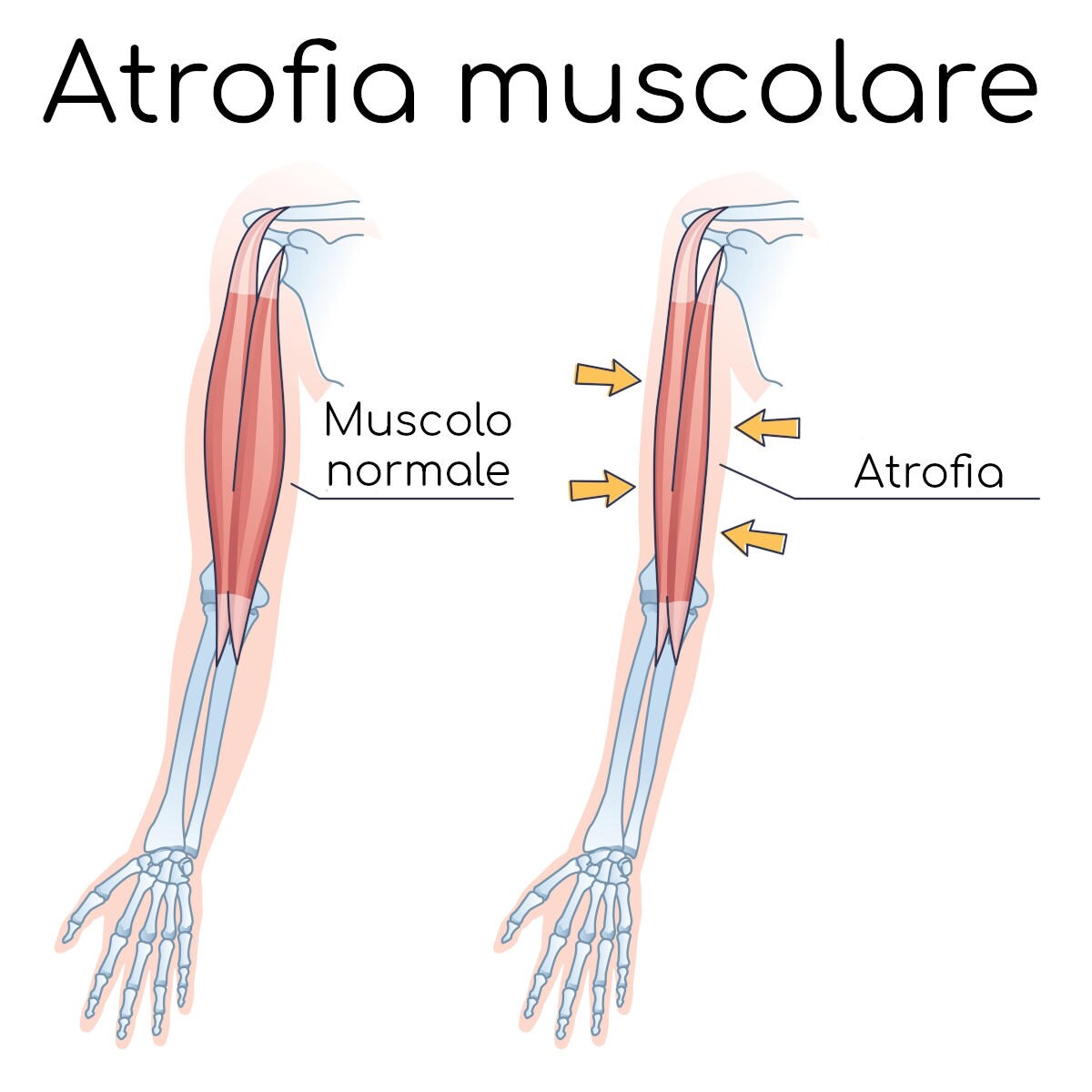
Esistono diverse forme di atrofia muscolare spinale, ma tutte accomunate da un esordio clinico usualmente infantile o giovanile con atrofia generale dei muscoli e conseguente deficit di forza, che si manifesta in forma di gravi difficoltà ad eseguire i movimenti più importanti come il semplice stare seduti o il riuscire a camminare.
Shutterstock/VectorMine
La malattia è causata da una mutazione genetica a carico dei geni SMN 1 o 2, con conseguente produzione alterata o ridotta di una proteina necessaria alla sopravvivenza dei motoneuroni.
La SMA è una malattia fortunatamente molto rara, con un’incidenza di circa 1 caso all’anno ogni 10.000 nuovi nati, che si trasmette generalmente in forma ereditaria.
Il quadro clinico è caratterizzato da una serie di sintomi neurologici, tra cui si annoverano:
- ritardo nello sviluppo motorio
- difficoltà nella suzione (che fa da ostacolo ad una corretta e adeguata nutrizione)
- disfagia con rischio di sviluppo di polmonite “ab ingestis”
- respiro debole di tipo diaframmatico
- deformità delle articolazioni, di solito poco stabili e funzionali
- incapacità di acquisire la capacità di stare seduto e di deambulare autonomamente
- polmonite ed insufficienza respiratoria nelle fasi tardive, che rappresentano spesso le cause del precoce exitus.
Il sospetto diagnostico dell’atrofia muscolare spinale viene posto dal medico sulla base dell’esame obiettivo neurologico e su alcuni dati anamnestici (come la presenza di storia familiare di SMA), ma la diagnosi di certezza avviene mediante test genetico su prelievo di sangue.
L’exitus avviene di solito in giovane età, spesso nei primi anni di vita, quando per il deficit dei muscoli respiratori si presentano sempre più spesso episodi di insufficienza respiratoria complicati da polmonite sempre meno gestibile con la terapia medica.
La prognosi della SMA, per questi motivi, è tendenzialmente infausta e sfavorevole.
Gli obiettivi principali delle cure consistono nell’alleviare i sintomi e i disturbi più spiacevoli, riconoscere e gestire le complicanze che si presentano nel tempo, cercando di mantenere il più possibile una qualità di vita accettabile.
Richiami di anatomia
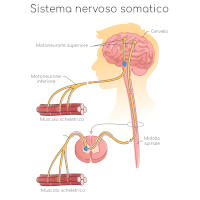
I motoneuroni sono cellule nervose che originano dal midollo spinale, all’interno della colonna vertebrale, e tramite lunghi prolungamenti (assoni) controllano l’attività dei muscoli, permettendone il trofismo (nutrizione) e la contrazione.
La SMA è una patologia che colpisce quasi esclusivamente i motoneuroni spinali, caratterizzata dalla perdita progressiva di quest’ultimi con conseguente atrofia muscolare e deficit di forza; coinvolge cioè i motoneuroni che originano dal midollo spinale e non quelli che originano dall’encefalo.
I motoneuroni spinali sono, per inciso, quelli addetti al controllo dei muscoli scheletrici (volontari) e dei muscoli lisci (involontari) del cuore e dei visceri.
Cause
La causa dell’atrofia muscolare spinale in tutte le sue forme è una mutazione genetica che nel 95% dei casi colpisce il gene SMN 1 (Survival Motor Neuron 1), che si trova sul cromosoma 5.
Questo gene normalmente codifica per una proteina coinvolta nel processo di apoptosi cellulare (morte programmata) ma che presenta anche funzioni di regolazione della trascrizione del DNA.
A seconda del tipo di mutazione si manifestano diverse forme cliniche di atrofia muscolare spinale.
Più raramente è possibile riscontrare mutazioni alleliche a livello del gene SMN 2, un gene modificatore con funzionalità strettamente connesse all’SNM 1.
In generale entrambi i geni SMN 1 e 2 servono a garantire la sopravvivenza dei motoneuroni e la loro mutazione porta ad una graduale degenerazione dei motoneuroni spinali, provocando atrofia muscolare da mancato utilizzo delle fibre muscolari che nei casi più severi porta ad una vera e propria paralisi.
Quando questa paralisi coinvolge i muscoli respiratori sopraggiunge una grave insufficienza respiratoria che si traduce nell’exitus del soggetto affetto.
Trasmissione genetica
Nel 99% dei pazienti è una patologia ereditaria, trasmessa dai genitori alla prole mediante un modello autosomico recessivo; affinché la malattia si manifesti è cioè necessario che entrambi i genitori siano portatori sani (eterozigoti) del difetto genetico e che entrambi trasmettano tale mutazione al figlio. Nella maggior parte dei casi i genitori, non manifestando sintomi, sono inconsapevoli della condizione.
Se nella coppia un solo genitore è portatore del gene mutato gli esiti possibili di ciascuna gravidanza sono i seguenti:
- 50% di concepimento di un figlio perfettamente sano
- 50% di concepimento di un figlio portatore sano
- 0% di concepimento di un figlio malato.
Nel caso di una coppia in cui entrambi i genitori siano portatori sani le probabilità per ciascuna gravidanza sono le seguenti:
- 25% di concepimento di un figlio perfettamente sano
- 50% di concepimento di un figlio portatore sano
- 25% di concepimento di un figlio malato.
Solo nell’1% dei casi la SMA può presentarsi come forma non ereditaria per la presenza di una mutazione “de novo” (casuale e imprevedibile) che si verifica in una fase molto precoce dello sviluppo dell’embrione.
Classificazione
Clinicamente le atrofie muscolari spinali possono essere classificate in diversi tipi:
- Tipo 0, la forma più grave che si manifesta già a livello fetale con sopravvivenza di poche settimane dalla nascita (con supporto respiratorio obbligato).
- Tipo I (Malattia di Werdnig-Hoffmann), caratterizzata da un’età di esordio inferiore ai 6 anni, con i bambini che non riusciranno ad acquisire la capacità di sedersi. Si tratta della forma certamente più grave con età del decesso di solito inferiore ai 2 anni ed anche la più frequente (circa metà dei casi).
- Tipo II, esordisce entro i 18 mesi nella maggior parte dei casi con i soggetti affetti che non riusciranno ad acquisire la capacità di camminare correttamente; il decesso sopraggiunge solitamente dopo i 2 anni di età.
- Tipo III (malattia di Kugelberg-Welander), presenta un esordio clinico dopo i 18 mesi con i soggetti giovani-adulti che questa volta riusciranno a restare in piedi o a deambulare autonomamente senza ausilio. Il decesso avviene quasi sempre in età adulta.
- Tipo IV, rappresenta una forma adulta della malattia ed anche la meno grave con esordio intorno ai 30 anni e un decorso clinico molto lento, con aspettativa di vita quasi normale.
- Atrofie muscolari spinali focali, forme molto rare caratterizzate da un’atrofia muscolare che questa volta è focalizzata (limitata) in pochi distretti senza essere generalizzata.
- atrofia spinale scapolo-peroneale,
- atrofia spinale monomelica, la cui variante più frequente è conosciuta come “malattia di Hirayama”,
- paralisi bulbare progressiva di Fazio-Londe.
Sintomi
Il quadro clinico dell’atrofia muscolare spinale tende a presentare sintomi neurologici sovrapponibili, ma con una certa differenza dovuta soprattutto alla diversa età di esordio della patologia.
L’atrofia muscolare spinale di tipo I e tipo II sono anche conosciute come “malattia di Werdnig-Hoffman”, con esordio in epoca pre-natale o subito dopo la nascita. Il neonato presenta sin da subito una ipotonia generalizzata, ovvero un tono muscolare molto ridotto che è percepibile quando lo si tiene in braccio. Questo sintomo di ipotonia caratteristica molto marcata viene chiamato “floppy infant” (volendo tradurre in italiano si parla di “bambino di pezza”).
Oltre all’ipotonia si riconoscono presenti altri segni e sintomi, perlopiù neurologici:
- ritardo nello sviluppo motorio
- deficit della suzione con difficoltà a nutrirsi e quindi ritardo di crescita
- disfagia, ovvero difficoltà alla deglutizione, che favorisce lo sviluppo di polmoniti “ab ingestis” molto gravi
- respiro prettamente di tipo diaframmatico per la debolezza dei muscoli intercostali
- assenza dei riflessi propriocettivi generalizzati
- assunzione della posizione “a rana”, ovvero con flessione e rotazione esterna delle cosce, con gomiti flessi ed intraruotati
- deformità delle articolazioni, di solito poco stabili e funzionali
- incapacità di acquisire la capacità di stare seduto e di deambulare autonomamente.
Nell’atrofia muscolare spinale di tipo II, l’esordio avviene di solito nei primi 2 anni di vita e il decorso clinico è più lento ma altrettanto ingravescente. Il decesso di solito sopraggiunge tra i 2 e i 10 anni.
Il quadro clinico è caratterizzato anche in questo caso da un marcato deficit motorio con atrofia muscolare, soprattutto a livello dei cingoli scapolare (spalla) e pelvico (bacino).
Anche in questo caso il bambino non riuscirà mai a deambulare senza ausilio, pur avendo una minima capacità di mantenere la posizione seduta.
Il decorso clinico diviene sempre più ingravescente e gravato da complicanze sempre meno gestibili come la polmonite o l’insufficienza respiratoria, che nel corso del tempo saranno le maggiori responsabile del precoce exitus.
L’atrofia muscolare spinale di tipo III è anche detta “malattia di Kugelberg-Welander”; presenta un esordio più tardivo rispetto alle forme precedenti ma comunque in età giovanile.
I sintomi principali di questa forma sono:
- deficit di forza muscolare
- atrofia della muscolatura dei cingoli
- presenza di scoliosi o di altre deformazioni della colonna
- capacità di deambulare ma con andatura anserina (ovvero simile a quella dell’oca quindi con
- dondolio laterale, di solito dovuta ad atrofia dei muscoli glutei).
Il decorso della SMA di tipo III è lentamente ma progressivamente invalidante, con riduzione notevole della qualità di vita, ma con solo una modesta riduzione della spettanza di vita.
Sono presenti anche in questo caso difficoltà a mantenere l’equilibrio, ad alzarsi da una posizione seduta e a camminare.
Atrofie muscolari spinali focali
- L’atrofia spinale scapolo-peroneale è una variante che si presenta con sintomi localizzati a spalla e braccio (si osserva atrofia dei muscoli delle spalle e del perone, l’osso compreso tra spalla e gomito, delle mani e dei pettorali).
- La malattia di Hirayama presenta atrofia progressiva monolaterale (solo un lato) dei muscoli della mano e dell’avambraccio e interessa quasi esclusivamente il sesso maschile.
- La paralisi bulbare progressiva di Fazio-Londe è una forma caratterizzata da un esordio nella prima infanzia che si presenta con sintomi quali:
- disfagia
- disartria, ovvero incapacità di articolare le parole in maniera corretta, con linguaggio che si presenta irregolare e difficilmente interpretabile
- atrofia del muscolo della lingua
- deficit della muscolatura mimica del volto
- deficit generalizzato dei muscoli (tardivamente).
Diagnosi
Il percorso diagnostico inizia con l’anamnesi, che consiste nel formulare una serie di specifiche domande che permettono al medico di ricostruire la storia clinica personale e familiare dell’ammalato; ovviamente nel caso delle atrofie muscolari spinali saranno principalmente i genitori a dover descrivere al medico i sintomi riscontrati.
I primi sintomi di allarme del neonato-bambino che devono essere valutati sono:
- l’incapacità del bambino di assumere la posizione da seduto o di mantenerla
- la difficoltà alla suzione e al nutrimento giù durante l’allattamento
- la respirazione difficoltosa e diaframmatica
- i forti problemi con l’inizio della deambulazione
- una muscolatura più debole e meno tonica
Trattandosi di forme genetiche ereditarie è importante indagare sulla presenza in famiglia di altri individui affetti da SMA o da patologie simili.
L’esame obiettivo neurologico deve essere quanto mai completo e di solito mette in evidenza i sintomi già descritti, come l’atrofia muscolare con deficit di forza soprattutto della muscolatura dei cingoli o l’assenza di riflessi propriocettivi.
Agli esami ematochimici è possibile riscontrare un aumento della creatina chinasi sierica (CK). Eventuali altre anomalie possono essere secondarie ad una malnutrizione conseguente alla difficoltà di deglutizione dei soggetti affetti.
All’elettromiografia (EMG) si evidenzia, in tutte le forme di SMA, un danno neurogeno mielopatico, con danni quindi alla guaina mielinica che riveste i nervi e ai nervi stessi.
La biopsia muscolare come l’elettromiografia possono dimostrare un danno muscolare di origine nervosa con danni della mielina.
Con il riscontro dei segni e dei sintomi tipici della malattia è possibile ipotizzare la presenza di una malattia neurologica, ma per la diagnosi di conferma sarà necessario eseguire un test genetico su un campione di sangue.
Sarà infine opportuno rivolgersi a figure specializzate come un Pediatra o un Genetista per impostare il corretto percorso diagnostico-terapeutico e giungere alla diagnosi specifica di SMA.
Con il test genetico è importante risalire anche alla sottoforma di atrofia muscolare spinale presente, per poter inquadrare il soggetto affetto nella evoluzione della malattia e prevederne i possibili sviluppi a breve termine.
In casi selezionati è possibile eseguire il test anche in fase prenatale, mediante villocentesi o amniocentesi; tale opportunità è riservata in genere alle donne che abbiano pianificato una gravidanza e abbiano già un figlio affetto da SMA, siano portatrici sani della malattie (o lo sia il loro partner) o abbiano una storia familiare di SMA.
Cura
Trattandosi di una grave patologia di origine genetica, purtroppo non esiste ad oggi una cura che ne permetta la guarigione; lo scopo del trattamento sarà perciò quelli di alleviare il più possibile i sintomi e controllare, gestire nel tempo le complicanze che si presentano.
Uno dei supporti principali di cui necessitano i soggetti affetti da SMA è quello alla locomozione. Anche nelle forme ad esordio tardivo diventa fondamentale il progressivo utilizzo di:
- stampelle
- carrelli per la deambulazione
- sedia a rotelle.
Nelle fasi tardive della malattia sopraggiunge la paralisi, con i soggetti che sono costretti all’allettamento permanente. In tali casi sono utili tutti i presidi per evitare la comparsa delle piaghe da decubito, come ad esempio specifici materassini antidecubito.
Il supporto respiratorio è necessario sin dalle prime fasi alla malattia, sia per evitare l’insufficienza respiratoria, sia per ridurre il rischio di infezioni respiratorie e di polmonite.
Nelle prime fasi saranno necessari supporti per la ventilazione non invasiva come naselli o maschere facciali, mentre nelle fasi più avanzate con autonomia respiratoria ormai perduta, diventa fondamentale il supporto ventilatorio invasivo mediante intubazione oro-tracheale.
Per alleviare il disturbo della malnutrizione e ridurre il rischio di polmonite “ab ingestis”, può essere necessario in varie fasi della malattia ricorrere alla nutrizione enterale mediante sondino naso-gastrico o mediante gastrostomia.
La fisioterapia permette di migliorare nei limiti del possibile la flessibilità dei muscoli e delle articolazioni, senza chiaramente poter risolvere l’ingravescente deficit di forza muscolare.
Negli ultimi anni si è assistito allo sviluppo di alcuni farmaci specifici contro la SMA, che riescono ad incrementare la produzione di proteina SMN e a mantenerne livelli adeguati; nonostante si prospettino buoni risultati, il loro limite attuale è il costo molto elevato. Si tratta in ogni caso di farmaci sintomatici, ovvero che riducono la gravità dei sintomi e non sono in grado di curare la malattia di base.
Una nuova frontiera terapeutica può essere quella della terapia genica che prevederebbe tecniche che permettono di correggere la mutazione genica responsabile della SMA, ma tali cure sono ancora in fase sperimentale.
Fonti e bibliografia
- Il Bergamini di Neurologia-Mutano, Lopiano , Durelli. Ed: Cortina (Torino).
- Spinal muscular atrophy – Adele D’Amico, Eugenio Mercuri, Francesco D Tiziano & Enrico Bertini
- MedScape
Tutti gli aggiornamenti su salute, alimentazione e benessere.