Introduzione
La Sclerosi Laterale Amiotrofica, o SLA, è una grave malattia neurologica che colpisce la capacità di muoversi; è anche chiamata malattia di Lou Gehrig, dal nome di un un giocatore di baseball statunitense colpito dalla malattia negli anni ’30 del secolo scorso.
In Italia si stima che ci siano circa 3500 malati di SLA e sono più di 1.000 le nuove diagnosi in un anno.
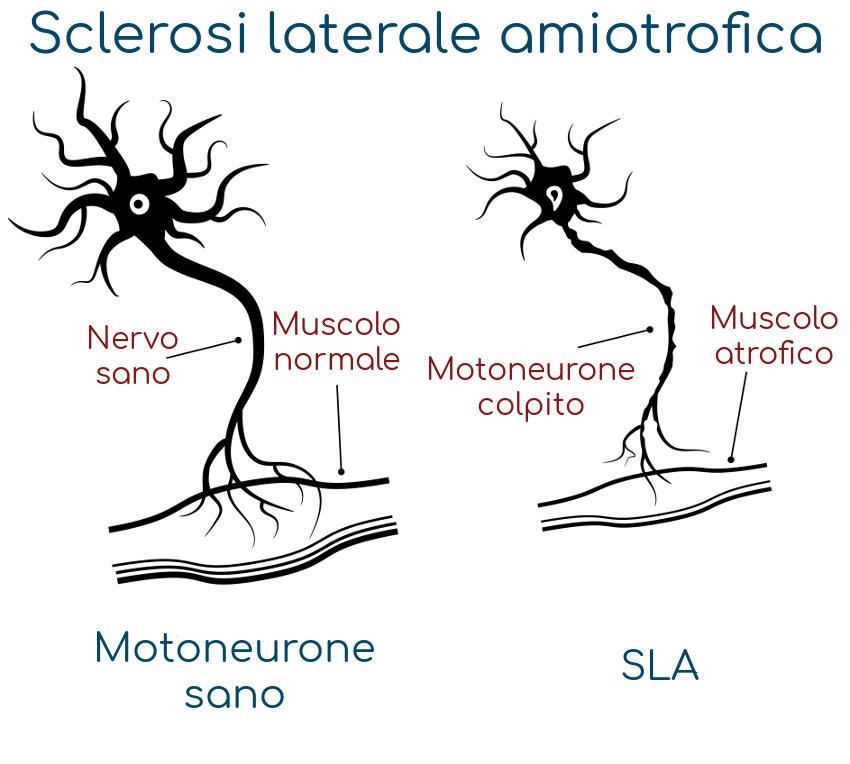
La sclerosi laterale amiotrofica è una malattia del sistema nervoso che attacca specifici neuroni (i motoneuroni, ossia le cellule nervose che si occupando dei movimenti) di cervello e midollo spinale; il compito di queste cellule è quello di trasmettere messaggi dal cervello e dal midollo spinale ai muscoli volontari, ossia quelli che si possono controllare e comandare, come quelli responsabili di braccia e gambe. In un primo momento questo è causa di problemi muscolari lievi e alcuni pazienti notano sintomi iniziali come
- difficoltà a camminare o correre,
- difficoltà a scrivere,
- difficoltà a parlare.
Con il progredire della malattia si perde completamente la forza e la capacità di movimento; quando ad essere colpiti sono infine i muscoli del torace, anche la respirazione può diventare difficoltosa prima e impossibile poi, rendendo indispensabile il supporto respiratorio fornito da speciali macchine.
La malattia di solito colpisce tra i 40 e i 60 anni e più frequentemente gli uomini che le donne; non si conoscono ancora le cause esatte della malattia, potrebbe esserci una qualche forma di famigliarità, ma più spesso sembra colpire a caso. La SLA non è contagiosa.
Purtroppo ad oggi non c’è cura: i farmaci possono alleviare i sintomi e, a volte, prolungare la sopravvivenza, ma non permettono la guarigione.
La maggior parte delle persone con diagnosi di sclerosi laterale amiotrofica muore per insufficienza respiratoria, di solito entro 3-5 anni dalla prima comparsa dei sintomi; circa il 10% dei pazienti riesce invece a sopravvivere per 10 o più anni.
Sebbene l’aspettativa media di vita di una persona con SLA sia da due a cinque anni dal momento della diagnosi, il decorso della malattia è variabile e molte persone possono convivere con la condizione molto più a lungo, come per esempio ha dimostrato il fisico Stephen Hawking:
- il venti percento delle persone con SLA vive cinque anni dal momento della diagnosi,
- il 10 percento dieci anni,
- il cinque percento vivrà 20 anni o più.
Anatomia
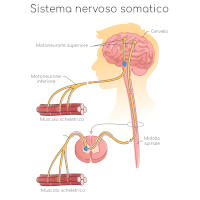
Il cervello e il midollo spinale costituiscono il fondamento del sistema nervoso centrale; i nervi di braccia, gambe, petto e qualsiasi altra parte del corpo costituiscono invece il sistema nervoso periferico.

In giallo il sistema nervoso centrale, in azzurro quello periferico – Di Medium69, Jmarchn – File:Nervous system diagram.png, CC BY-SA 4.0, Collegamento
Il cervello è simile a un computer molto complesso in grado di:
- raccogliere i segnali provenienti dalla periferia del corpo attraverso i nostri sensi,
- comandare i muscoli per svolgere azioni più o meno complesse.
Le cellule che costituiscono il sistema nervoso centrale sono diverse da tutte le altre e, insieme, forniscono la capacità di elaborare le informazioni raccolte e rispondere di conseguenza; per farlo sono collegate in modo fitto tra di loro a formare una complessa e sofisticata rete neurale (le cellule nervose prendono il nome di neuroni ), comunicando attraverso filamenti detti assoni che sono in grado di trasmettere impulsi elettrici.
I neuroni che controllano i nostri muscoli sono chiamati motoneuroni: gli assoni dei motoneuroni fanno contrarre i nostri muscoli e sono quindi responsabili di tutti i movimenti e delle azioni che siamo in grado di compiere: dall’alzare un dito al cantare, dal saltare allo scrivere a PC, dal ridere al fare un tuffo.
La SLA colpisce i neuroni che controllano i muscoli, i motoneuroni.
I messaggi provenienti dai motoneuroni del cervello, chiamati motoneuroni superiori, sono trasmessi ai motoneuroni del midollo spinale, chiamati motoneuroni inferiori, poi sono trasmessi ai muscoli.
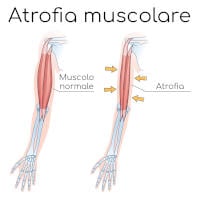
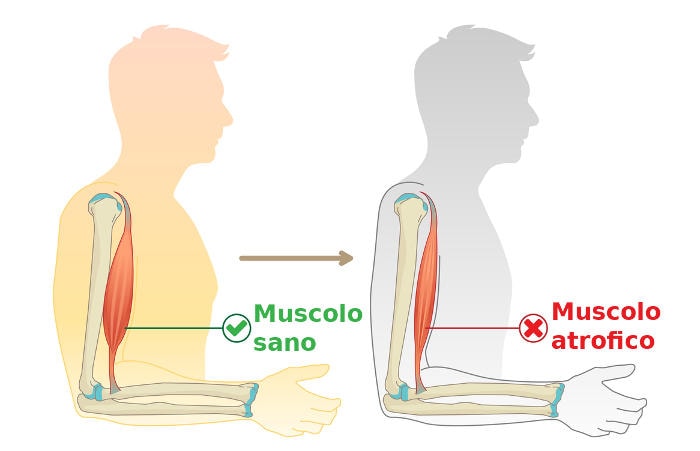
Nella SLA sia i motoneuroni superiori che quelli inferiori vanno incontro a perdita di funzionalità fino alla completa morte, smettendo così di inviare messaggi ai muscoli; impossibilitati a funzionare i muscoli gradualmente si indeboliscono, iniziano a muoversi involontariamente (fascicolazioni) e perdono tono e massa (atrofia). Alla fine il cervello perde la sua capacità di avviare e controllare i movimenti volontari.
iStock.com/leremy
La malattia è progressiva, quindi i danni evolvono gradualmente e così i sintomi relativi.
Cause
La causa di della SLA è tuttora sconosciuta, anche se si ritiene che si tratti di una combinazione di fattori genetici e ambientali.
Un importante passo verso la determinazione dei fattori di rischio della sclerosi laterale amiotrofica è stato effettuato nel 1993, quando si è individuata una mutazione nel DNA (gene SOD1) associata ad alcuni casi di SLA familiare. Sebbene non sia ancora chiaro il legame tra mutazione e degenerazione dei motoneuroni, è sempre più evidente che il gene svolge un ruolo determinante nella produzione di proteine mutanti con la capacità di causare danni alle strutture nervose.
Da allora sono state identificate più di una dozzina di mutazioni genetiche aggiuntive, con effetti e ripercussioni diverse, ma con la conseguenza invariabile di creare problemi alla trasmissione del segnale nervoso.
Una seconda area di ricerca riguarda lo studio dell’impatto dei fattori ambientali, quali per esempio
- esposizione ad agenti tossici o infettivi,
- virus,
- traumi fisici,
- dieta,
- fattori comportamentali e professionali.
Esistono per esempio ipotesi relative al possibile innesco della malattia causato dall’esposizione a sostanze tossiche (piombo, pesticidi, …) durante la guerra o a un eccesso di attività fisica, fattori che potrebbero spiegare l’aumento di frequenza di comparsa della SLA tra i reduci e gli atleti.
Fattori di rischio
La malattia può colpire qualunque persona senza distinzione di razza o etnia, ma si è notato che
- nonostante la SLA possa colpire a qualunque età, sembra essere particolarmente diffusa tra i 55 e i 75 anni.
- gli uomini sono leggermente più colpiti rispetto alle donne.
Alcune fonti identificano il fumo come possibile fattore di rischio ambientale, mentre c’è maggior consenso relativamente all’esposizione professionale a tossine, agenti chimici e, secondo alcuni ricercatori, un eccesso di attività fisica.
Più del 90% dei casi di SLA colpisce in modo sporadico, ossia senza associazioni visibili per fattori di rischio o famigliarità.
Circa il 5-10% dei casi sono invece legati a una qualche forma di famigliarità.
Sintomi
L’insorgenza della SLA è spesso così graduale che i sintomi vengono trascurati per diverso tempo, fino quando atrofia e debolezza non diventano così evidenti da instillare il dubbio al medico.
I sintomi iniziali della SLA includono:
- fascicolazioni (contrazioni muscolari rapide e involontarie) nel braccio, nella gamba, nella spalla o nella lingua,
- crampi muscolari,
- rigidità muscolare (spasticità),
- debolezza muscolare che colpisce un braccio, una gamba, un collo o il diaframma,
- difficoltà di linguaggio e voce nasale,
- difficoltà a masticare e/o deglutire.
Per molti soggetti il primo segno può comparire a livello di mano braccio, a seguito di difficoltà con compiti semplici come abbottonare una camicia, scrivere o inserire una chiave nella serratura.
In altri casi i sintomi inizialmente colpiscono una delle gambe e i pazienti incontrano quindi difficoltà a camminare, correre o si rendono conto di inciampare più spesso.
Altri individui, infine, notano prima difficoltà di linguaggio o di deglutizione.
Indipendentemente da dove i sintomi si manifestano per la prima volta, la debolezza muscolare e l’atrofia si diffondono progressivamente in altre parti del corpo, manifestandosi sotto forma di problemi di
Alcuni pazienti manifestano alterazioni cognitive, che si manifestano con lo sviluppo di reazioni inappropriate al contesto (sorriso, riso, pianto, …).
Anche la progressione dei sintomi non è sempre lineare; non è raro andare incontro a periodi, di durata variabile tra qualche settimana ed alcuni mesi, in cui non si verificano peggioramenti.
Sebbene la sequenza di emergenza dei sintomi e la velocità di progressione della malattia varino da persona a persona, alla fine il paziente invariabilmente perderà la capacità di stare in piedi o camminare, entrare o uscire da solo dal letto o utilizzare mani e braccia.
Le difficoltà di deglutizione e masticazione sono causa di aumento del rischio di soffocamento, inoltre il soggetto affetto da SLA brucia calorie a un tasso più rapido della norma e per tutti questi motivi la tendenza comune è quella di perdere peso rapidamente e andare incontro a malnutrizione.
Di norma rimane inalterata la capacità di ragionamento e la memoria, quindi la consapevolezza della progressiva perdita di funzione è un fattore predisponente disturbi quali ansia e depressione.
Solo una una piccola percentuale di individui affetti da SLA potrebbero sviluppare una forma di demenza nelle fasi più avanzate della malattia.
Con l’indebolimento dei muscoli del sistema respiratorio aumentano le difficoltà di respirazione, che termina purtroppo con la necessaria dipendenza da una macchina che possa supportare la respirazione. Aumenta il rischio di polmonite durante le fasi successive e, oltre alla presenza di i crampi muscolari che possono causare disagio, alcuni individui con sclerosi laterale amiotrofica possono sviluppare una dolorosa neuropatia.
Diagnosi
La diagnosi di Sclerosi Laterale Amiotrofica rappresenta ancora oggi una sfida clinica complessa, poiché non esiste un singolo test biochimico o strumentale in grado di confermare la patologia con certezza assoluta. Il percorso diagnostico si basa sull’integrazione di evidenze cliniche, indagini elettrofisiologiche e l’esclusione di altre patologie che possono mimare i sintomi della SLA (diagnosi differenziale).
Il protocollo diagnostico attuale segue criteri internazionali aggiornati, come i Criteri di Gold Coast, che semplificano il processo focalizzandosi sulla presenza di segni di danno ai motoneuroni superiori e inferiori in una o più regioni del corpo, con carattere progressivo.
Esame clinico e neurologico
Il primo passo è una valutazione neurologica approfondita. Il medico ricerca segni di compromissione del primo motoneurone (come spasticità e riflessi patologici) e del secondo motoneurone (come atrofia muscolare e fascicolazioni). L’osservazione della progressione dei sintomi nel tempo è fondamentale per confermare il sospetto clinico.
Indagini elettrofisiologiche
L’elettromiografia (EMG) rimane il pilastro fondamentale della diagnosi. Questo esame permette di:
- Rilevare l’attività elettrica muscolare che indica la denervazione in corso (sofferenza del motoneurone inferiore).
- Documentare il coinvolgimento di muscoli clinicamente ancora sani, dimostrando la diffusione della malattia.
- Distinguere la SLA da altre malattie dei nervi o dei muscoli.
L’elettroneurografia viene invece utilizzata per valutare la velocità di conduzione dei nervi sensitivi e motori, aiutando a escludere neuropatie infiammatorie o compressive.
Biomarcatori e test genetici
Una delle innovazioni più significative riguarda l’uso dei biomarcatori. Il dosaggio dei neurofilamenti (catena leggera dei neurofilamenti, NfL) nel sangue o nel liquido cerebrospinale è diventato uno strumento prezioso per supportare la diagnosi e valutare la rapidità di progressione della malattia. Elevati livelli di NfL indicano un danno assonale attivo.
Il test genetico è oggi raccomandato non solo nei casi con familiarità nota, ma sempre più spesso anche nelle forme sporadiche. Identificare mutazioni in geni come SOD1, C9orf72, TARDBP o FUS è cruciale sia per la prognosi che per l’accesso a terapie geniche personalizzate di recente introduzione.
Imaging e altri esami
La risonanza magnetica (RMN) dell’encefalo e del midollo spinale viene eseguita principalmente per escludere altre condizioni, come tumori, ernie discali cervicali severe o sclerosi multipla, che potrebbero giustificare la sintomatologia motoria. Tecniche avanzate di RMN possono mostrare segni di degenerazione dei fasci piramidali, supportando ulteriormente il sospetto di SLA.
Cura e terapia
Gli obiettivi principali della terapia della SLA sono il rallentamento della progressione della malattia, la gestione dei sintomi e il mantenimento della migliore qualità di vita possibile. Sebbene non esista ancora una cura risolutiva, l’approccio terapeutico è radicalmente cambiato grazie alla medicina di precisione e a una gestione multidisciplinare più aggressiva.
Le opzioni terapeutiche includono:
- Terapie farmacologiche modificanti la malattia (disease-modifying).
- Terapie geniche per sottogruppi specifici.
- Trattamenti sintomatici.
- Supporto tecnologico (respiratorio e nutrizionale).
- Riabilitazione e cure palliative precoci.
Terapie farmacologiche e innovative
Il riluzolo rimane la terapia standard di prima linea per tutti i pazienti, agendo sulla modulazione del glutammato per rallentare il danno neuronale. Ad esso si è affiancato l’edaravone, somministrato per via endovenosa o orale, che agisce come antiossidante per contrastare lo stress ossidativo nei motoneuroni.
La vera frontiera è rappresentata dalle terapie a bersaglio molecolare. Per i pazienti con mutazione del gene SOD1, è oggi disponibile il tofersen, un oligonucleotide antisenso che riduce la produzione della proteina tossica, mostrando risultati senza precedenti nel rallentare la patologia e ridurre i livelli di neurofilamenti.
Gestione multidisciplinare e stile di vita
La gestione della SLA richiede un team esperto (neurologo, fisiatra, pneumologo, nutrizionista, logopedista e psicologo). Lo stile di vita gioca un ruolo determinante:
- Nutrizione: è fondamentale mantenere un elevato apporto calorico e proteico. La perdita di peso è strettamente correlata a una prognosi peggiore. In caso di disfagia severa, si ricorre alla PEG (gastrostomia endoscopica percutanea) per garantire nutrimento e idratazione in sicurezza, evitando polmoniti ab ingestis.
- Attività fisica: deve essere moderata e mirata a prevenire la contrattura muscolare e il dolore articolare, senza eccedere nello sforzo che potrebbe affaticare ulteriormente i motoneuroni residui.
Supporto respiratorio
Il monitoraggio costante della funzionalità respiratoria permette l’introduzione tempestiva della ventilazione non invasiva (NIV). Questo supporto, inizialmente notturno, migliora significativamente la qualità del sonno, riduce la stanchezza diurna e prolunga la sopravvivenza più di ogni farmaco tradizionale. L’uso di macchine per la tosse assistita aiuta a mantenere libere le vie aeree.
Trattamenti sintomatici
Esistono soluzioni efficaci per molti dei disturbi correlati:
- Sialorrea (eccesso di saliva): trattata con farmaci anticolinergici o infiltrazioni di tossina botulinica nelle ghiandole salivari.
- Labilità emotiva: gestita con specifici farmaci che controllano le crisi di riso e pianto inappropriati.
- Crampi e spasticità: trattati con miorilassanti.
- Umore e sonno: l’uso di antidepressivi e terapie per i disturbi del sonno è fondamentale per il benessere psicofisico, così come l’attenzione alla regolarità intestinale per prevenire la costipazione.
La pianificazione anticipata delle cure e il coinvolgimento precoce nelle cure palliative garantiscono che le scelte del paziente siano sempre rispettate durante l’evoluzione della malattia.
Fonti e bibliografia
Le domande più frequenti
Cos'è la SLA?
Quali sono i sintomi iniziali? Come ci si accorge della SLA?
Come si manifesta?
Quando compaiono le fascicolazioni?
Come si diagnostica?
Quali sono i sintomi comuni della Sclerosi Laterale Amiotrofica (SLA) e come si differenziano da disturbi comuni di ansia o stress?
Quando è consigliabile un consulto neurologico in caso di sintomi muscolari o neurologici?
La SLA è ereditaria e quali sono le probabilità di svilupparla se ci sono precedenti in famiglia?
Come si diagnostica la SLA e quali esami sono necessari?
Tutti gli aggiornamenti su salute, alimentazione e benessere.




{kind=link}