Introduzione
I sarcomi dei tessuti molli sono un gruppo di tumori rari che colpiscono i tessuti che connettono, supportano e circondano altre strutture e organi del corpo. I tessuti che possono essere colpiti dai sarcomi comprendono grasso, muscoli, vasi sanguigni, tessuti profondi della pelle, tendini e legamenti e possono svilupparsi in quasi tutte le parti del corpo, comprese le gambe, le braccia e la pancia (addome).
Il liposarcoma è una rara neoplasia che interessa il grasso profondo, tra cui quello presente a livello di
- cavità poplitea (dietro al ginocchio),
- retroperitoneo (regione addominale)
- ed esofago.
Il trattamento prevede in genere un intervento chirurgico di rimozione del tumore, ma possono talvolta essere previsti anche altri trattamenti, come la radioterapia.
Immagini
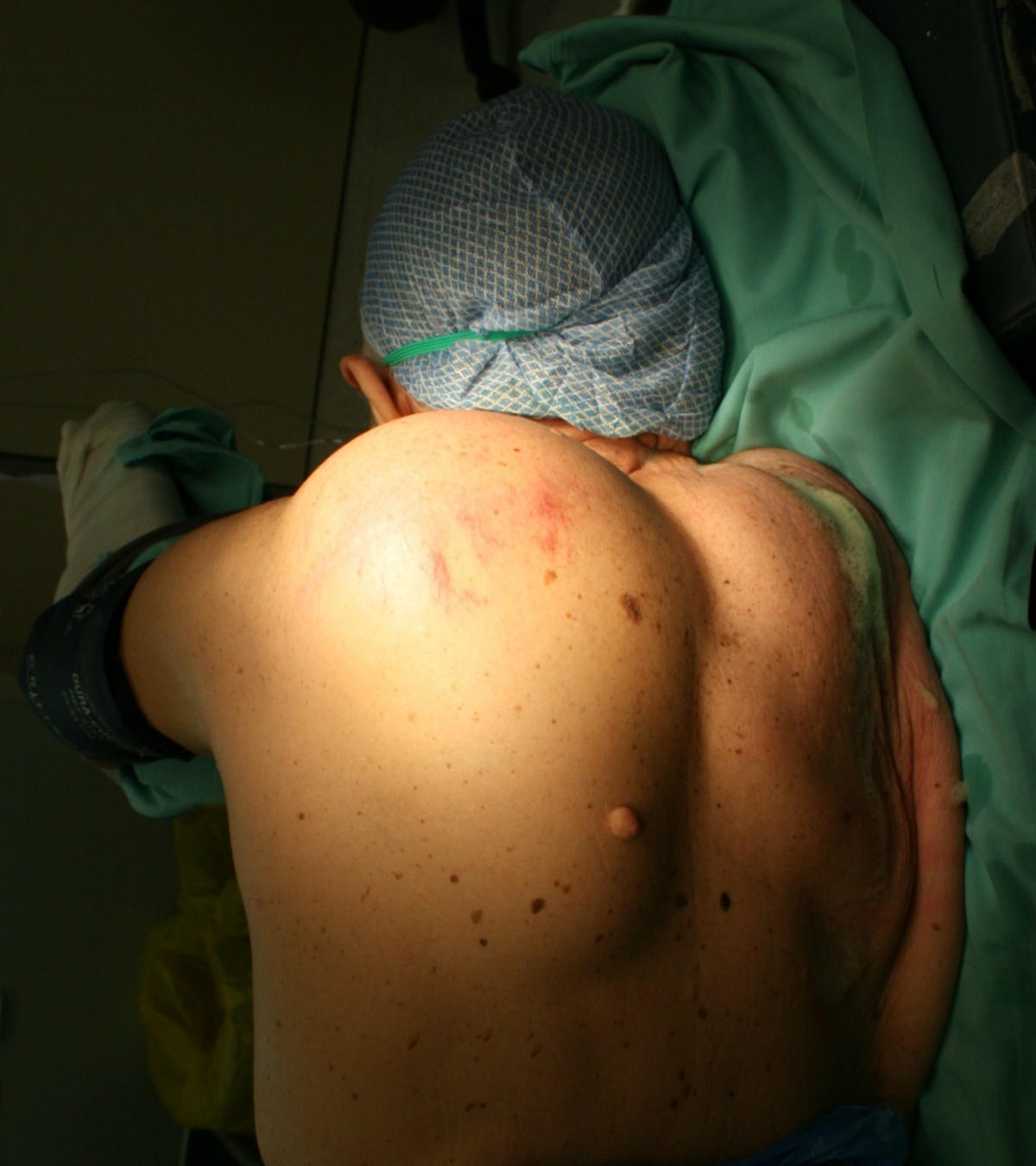
By Panoraia Paraskeva, Paraskevas Katsaronis, Eleftherios D Spartalis, Andreas C Lazaris, Hara Gakiopoulou, Panagiotis Mallis and Periklis Tomos – Panoraia Paraskeva, Paraskevas Katsaronis, Eleftherios D Spartalis, Andreas C Lazaris, Hara Gakiopoulou, Panagiotis Mallis and Periklis Tomos: Giant liposarcoma of the back with 4 types of histopathology: a case report In: Cases Journal 2009, 2:9339 doi:10.1186/1757-1626-2-9339 (Open Access), [1], CC BY-SA 2.0, https://commons.wikimedia.org/w/index.php?curid=10008970
È possibile vedere una seconda immagine di liposarcoma alla pagina seguente: https://en.wikipedia.org/wiki/File:Edemaliposarcoma.jpg
Sopravvivenza
Le statistiche relative alla sopravvivenza variano ovviamente in base allo studio e al Paese preso in considerazione, ma possono essere indicativamente schematizzate come segue (fonte cancer.org):
- Liposarcoma localizzato: 81%
- Diffusione regionale: 56%
- Diffusione a distanza: 15%
- Complessiva: 65%
La percentuale rappresenta il numero di pazienti (su 100) ancora vivi a 5 anni dalla prima diagnosi.
È possibile differenziale i dati in base alla localizzazione, trovando che:
- Quasi 90 persone su 100 con liposarcoma ben differenziato sopravvivono per 5 anni o più dopo la diagnosi.
- Quasi tutti i pazienti con liposarcoma ben differenziato delle braccia o delle gambe sopravvivono per 5 anni o più dopo la diagnosi.
- Circa 90 persone su 100 con liposarcomi ben differenziati nel tronco (torace, pancia e area pelvica) sopravvivono al cancro per 5 anni o più dopo la diagnosi.
- Circa 60 persone su 100 con liposarcoma ben differenziato dietro gli organi della pancia sopravvivono per 5 anni o più dopo la diagnosi.
È infine possibile valutare in base alla forma (le discrepanze sono legate a fonti differenti):
- liposarcoma ben differenziato è del 100% a 5 anni e dell’87% a 10 anni,
- liposarcoma mixoide 88% a 5 anni e al 76% a 10 anni,
- liposarcoma pleomorfofino 56% a 5 e e 39% a 10 anni.
Cause
Complessivamente i sarcomi dei tessuti molli sono tumori rari, che negli adulti italiani colpiscono circa 5 persone ogni 100.000 rappresentando l’1 per cento dei tumori maligni.
Negli Stati Uniti il liposarcoma rende conto di meno di un caso su 5 di sarcoma dei tessuti molli, di cui è comunque la forma più comune.
L’età media della diagnosi è di 50 anni, mentre non si rilevano differenze di etnia o sesso.
Come per la maggior parte dei tumori la causa è ancora sconosciuta, ma l’American Cancer Society ha identificato alcuni possibili fattori di rischio:
- esposizione a radiazioni (in particolare la radioterapia usata per trattare altri tumori maligni),
- alcune sindromi tumorali familiari (neurofibromatosi, sindrome di Gardner, retinoblastoma, sclerosi tuberosa, …),
- danni/traumi al sistema linfatico (ad esempio in caso di lesioni da radioterapia o in seguito alla rimozione di vasi linfatici, che possono condurre a linfedema e potenzialmente in seguito linfangiosarcoma),
- esposizione a sostanze chimiche tossiche.
È importante notare che:
- esistono casi in cui i pazienti non presentano alcun fattore di rischio per la malattia,
- i liposarcomi non si sviluppano dai lipomi, che sono invece fenomeni assolutamente benigni.
Classificazione
L’Organizzazione Mondiale della Sanità distingue 4 diversi sottotipi di liposarcoma, caratterizzati da specifiche alterazioni genetiche:
- ben differenziato (il più diffuso)
- mixoide (si localizza tipicamente a in braccia o gambe ed esordsisce generalmente a un’età inferiore rispetto alle altre forme)
- pleomorfo (raro, aggressivo, a progressione rapida; interessa i tessuti molli di gambe e braccia)
- dedifferenziato (il più pericoloso, tipicamente evoluzione dell sottotipo ben differenziato; fa la sua comparsa in addome, ma si diffonde rapidamente con metastasi a distanza).
Sintomi
Presentazione ed evoluzione del liposarcoma dipendono essenzialmente dalla posizione; la sede più comune è nelle estremità (gambe e braccia), poi retroperitoneo e più raramente l’esofago.
Il liposarcoma mixoide delle estremità si presenta come una massa profonda che interessa tipicamente la gamba.
La maggior parte dei pazienti colpiti da liposarcoma inizialmente non ha sintomi, che si sviluppano solo quando il tumore diventa abbastanza grande da esercitare un effetto di massa sulle strutture circostanti (premendo su muscoli, nervi, organi, …). A questo punto possono comparire:
- dolore,
- gonfiore (edema), in forma di pallina visibile esternamente,
- perdita funzionale e debolezza (legata alla compressione di nervi e vasi sanguigni).
Tra gli altri disturbi lamentati dal paziente figurano:
- alterazioni della sensibilità (come formicolio, pizzicore, solletico, prurito, sensazione di punture di spillo, …),
- vene varicose,
- affaticamento,
- perdita di peso involontaria,
- nausea e vomito.
Sono stati infine segnalati ulteriori sintomi sistemici come:
Il liposarcoma del retroperitoneo può crescere inosservato fino a stadi avanzati (fino a pesare diversi chili) a causa dell’aspecificità dei sintomi, che possono comprendere:
- dolore addominale ,
- gonfiore addominale,
- precoce senso di sazietà,
- stitichezza,
- presenza di sangue nelle feci.
Il sito più comune di liposarcoma esofageo è l’esofago superiore (l’esofago è il canale che collega la gola allo stomaco) e si presenta con
- difficoltà di deglutizione,
- perdita di peso.
Sono stati segnalati casi di masse così grande da arrivare a sporgere fin verso il cavo orale.
Diagnosi
Clinicamente può essere notato per la prima volta, soprattutto in caso di gambe e braccia, a seguito di traumi recenti, che favoriscono la scoperta della massa, ma causa (trauma) ed effetto (tumore) sono molto probabilmente del tutto casuali.
Al di là dei necessari passi introduttivi (raccolta di informazioni ed esame fisico), la diagnosi viene formulata principalmente mediante esami di imaging:
ed eventualmente gastroscopia.
La diagnosi di certezza, oltre che la caratterizzazione del tumore, è spesso ottenuta mediante biopsia (procedura che prevede il prelievo di un piccolo campione di tessuto tumorale per la successiva analisi di laboratorio); poiché molti di questi tumori si trovano in profondità, l’ecografia può essere particolarmente utile per guidare in tempo reale il medico durante l’introduzione dell’ago.
Cura
L’asportazione chirurgica è il cardine del trattamento, che tuttavia può risultare più o meno complessa in base alla localizzazione del tumore ed alla sua vicinanza con eventuali organi vitali.
Per le lesioni più avanzate potrebbe essere richiesta un’asportazione chirurgica ampia e profonda, eventualmente associata a radioterapia adiuvante (dopo l’intervento, per rimuovere ogni traccia di cellule tumorali e prevenire così recidive) e/o chemioterapia (che rimane tuttavia un approccio con scarsa letteratura a supporto).
Fonti e bibliografia
- Liposarcoma – Rabia Zafar; Yurong Wheeler
Tutti gli aggiornamenti su salute, alimentazione e benessere.






{kind=link}