Introduzione
Il termine neurofibromatosi si riferisce a un gruppo di malattie genetiche multisistemiche che causano la crescita di tumori lungo i nervi. Attualmente, la classificazione clinica distingue tre forme principali:
- neurofibromatosi di tipo 1 (NF1, la forma più diffusa),
- neurofibromatosi di tipo 2 (NF2, oggi spesso inquadrata nelle schwannomatosi correlate a NF2)
- e la schwannomatosi (o NF3).
Ognuna di esse è causata da una diversa mutazione genetica e caratterizzata da un diverso tipo di tumori delle guaine nervose e malformazioni di altri tessuti (cute, ossa ed altri organi). L’entità e la gravità delle lesioni variano da caso a caso, così come la prognosi.
Non esistono misure preventive per queste patologie. Alcuni centri specialistici dispongono di test di diagnosi prenatale per l’identificazione delle mutazioni genetiche responsabili durante la gravidanza.
Cause
La neurofibromatosi di tipo 1 (NF1), o malattia di von Recklinghausen, è dovuta alla mutazione del gene oncosoppressore NF-1, anche detto neurofibromina, una proteina coinvolta nel controllo della crescita cellulare che si trova sul cromosoma 17.
La metà dei casi è familiare, ovvero la mutazione genetica viene trasmessa da genitore a figlio con modalità autosomica dominante (il figlio di un individuo affetto ha la probabilità del 50% di essere anch’egli affetto); tuttavia capita spesso che un genitore affetto da neurofibromatosi riceva la diagnosi solo quando il figlio viene avviato ad una valutazione specialistica. La malattia ha infatti un’espressività variabile, ovvero può manifestarsi con segni clinici diversi anche all’interno di una stessa famiglia ed a diverse età. Gli altri casi “non familiari” sono dovuti ad una mutazione spontanea, ex novo, del gene neurofibromina: in questi casi un soggetto malato ha dei genitori sani.
La neurofibromatosi di tipo 2 (NF2), invece, è dovuta alla mutazione del gene NF-2, localizzato sul cromosoma 22, che codifica per una proteina oncosoppressiva chiamata merlina o schwannomina, anch’essa coinvolta nel controllo della crescita cellulare.
Anche nella neurofibromatosi di tipo 2, il 50% dei casi è familiare (trasmissione autosomico dominante), l’altro 50% è dovuto a mutazioni nuove, non ereditate.
La schwannomatosi è causata infine dalla mutazione del gene INI1/SMARCB1 o del gene LZTR1, che producono proteine deputate al controllo della proliferazione cellulare.
Sintomi
Neurofibromatosi di tipo 1
La neurofibromatosi di tipo 1 è la più comune neurofibromatosi, con una prevalenza di un caso su 3000. È caratterizzata dalla presenza di multipli neurofibromi (tumori benigni delle guaine dei nervi) e da manifestazioni cutanee, oculari ed ossee.
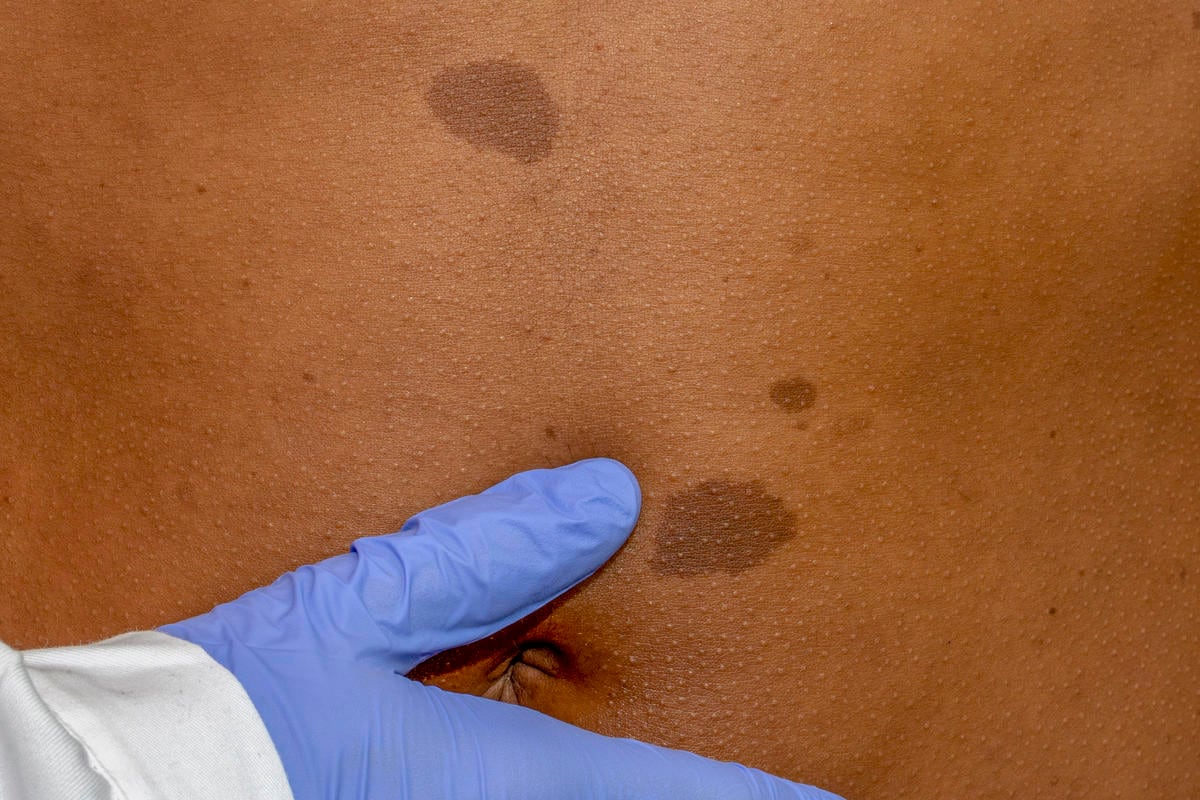
La maggior parte degli individui affetti sviluppa durante i primi anni di vita le cosiddette “macchie caffè latte”, ovvero delle lesioni piane di colore marrone chiaro che misurano almeno 5 mm prima della pubertà e 15 mm dopo la pubertà; il secondo segno in ordine cronologico è la lentigginosi delle pieghe, ovvero la comparsa di piccole macchie dapprima nelle regioni inguinali ,e poi ascellari, intorno ai 3-5 anni di età.
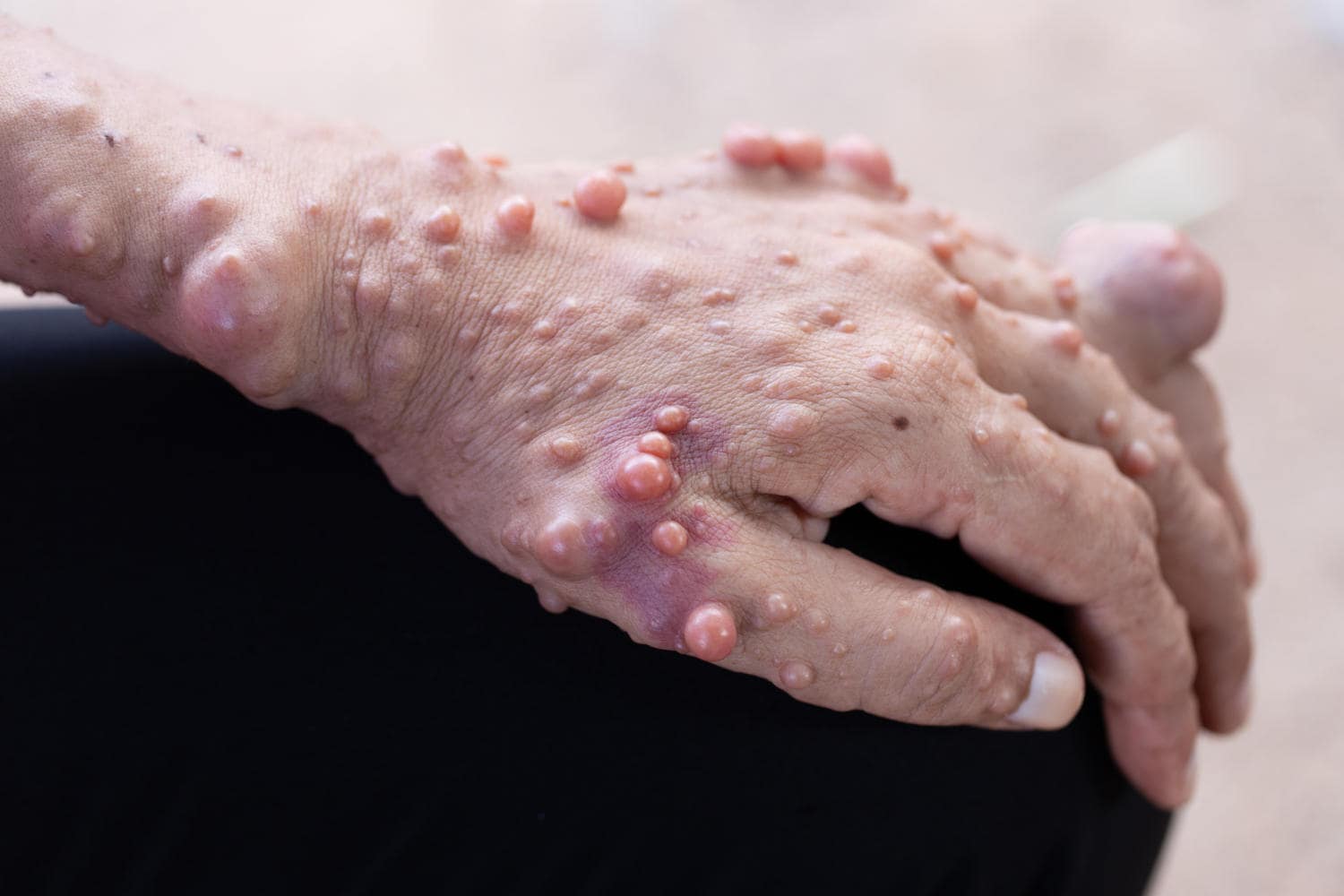
I neurofibromi sono tumori benigni che originano dalle guaine dei nervi: possono verificarsi sulla superficie cutanea, nel derma e negli organi interni. La loro insorgenza può avvenire durante l’infanzia, ma il numero tende aumentare specialmente dopo la pubertà.
I neurofibromi cutanei sono asintomatici, mentre quelli interni possono causare dolore o deficit neurologici, sulla base dei rapporti che instaurano con gli altri organi (rappresentano infatti delle masse occupanti spazio che comprimono gli organi vicini).
I noduli di Lisch sono tumori melanocitari dell’iride, altamente specifici della NF1, che compaiono di solito nella tarda infanzia. Sono visibili con una lampada a fessura (strumento a disposizione degli oculisti) in quasi tutti i pazienti dopo i 50 anni.
I gliomi ottici sono tumori che coinvolgono il 15% dei pazienti con NF1, specialmente a livello del nervo ottico: nella maggior parte dei casi sono asintomatici, ma possono anche interferire con la visione o causare disturbi neurologici.
Le anomalie ossee includono alterazioni delle ossa lunghe (tibia, radio, ulna) con curvatura degli arti oppure alterazioni delle ossa craniche come lo sfenoide.
Neurofibromatosi di tipo 2
La neurofibromatosi di tipo 2 (NF2) è molto meno frequente del tipo 1, verificandosi in un soggetto su 25000-60000. È caratterizzata dalla presenza degli schwannomi (detti anche neurinomi o neurilemomi) ovvero tumori benigni che compaiono in età infantile o adulta e costituiti da cellule che derivano dalla guaina dei nervi periferici. Tali shwannomi interessano entrambi i nervi vestibolari (nervi cranici che trasmettono al cervello le informazioni codificate a livello dell’orecchio e che sono deputati al senso dell’equilibrio e dell’udito): si manifestano dapprima con capogiri, vertigini, tinnito (sensazione di ronzio alle orecchie) ed in fase avanzata con sordità.
Altri tumori tipici sono gli schwannomi dei nervi cranici, spinali e periferici, i meningiomi intracranici, intraspinali ed alcuni tumori maligni del sistema nervoso centrale (ependiomi di basso grado).
I segni oftalmici dovuti all’interessamento dei nervi ottici comprendono una riduzione dell’acuità visiva e la cataratta.
Manifestazioni cutanee sono presenti solo in alcuni casi di NF2 sotto forma di macchie caffè-latte e schwannomi dermici (placche cutanee rosee o eritematose).
Neurofibromatosi di tipo 3
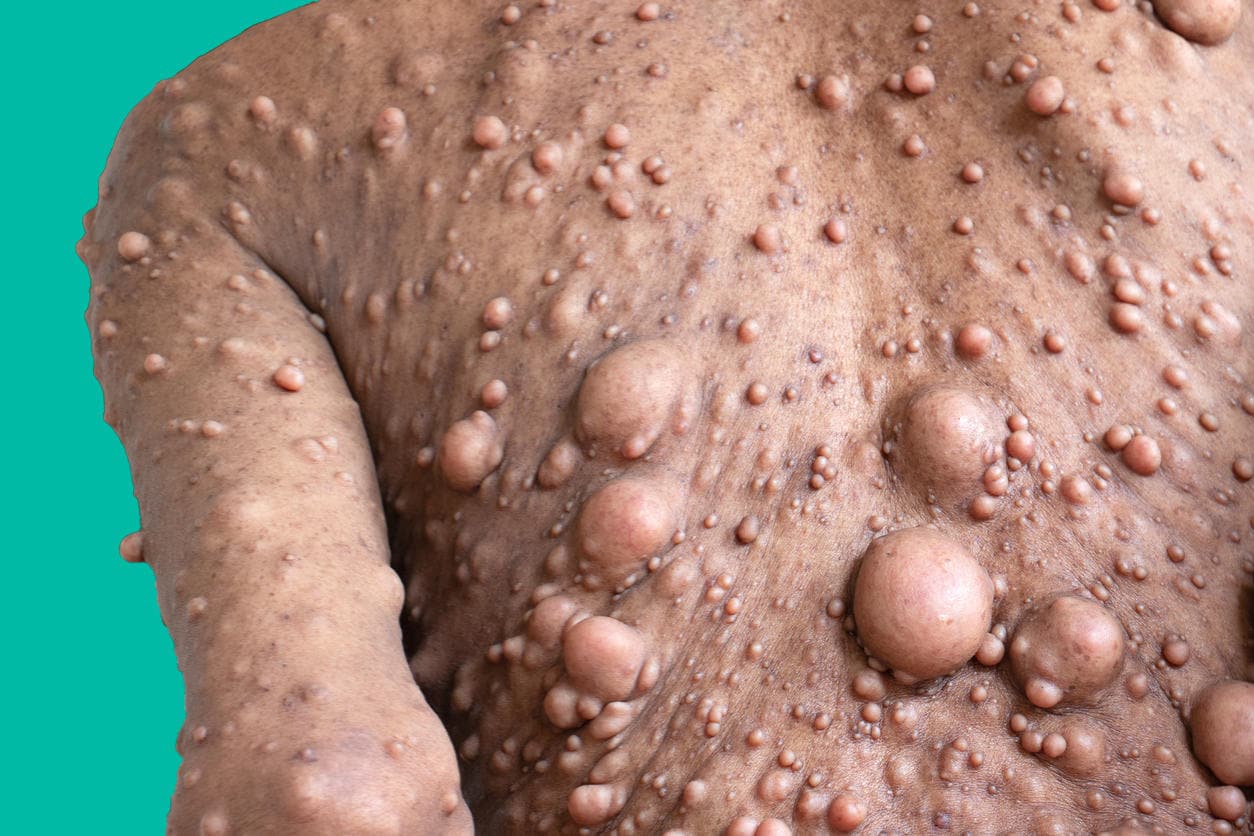
La neurofibromatosi di tipo 3 (NF3) o schwannomatosi è la malattia aggiunta più di recente al gruppo delle neurofibromatosi, pertanto i dati sulla prevalenza non sono ancora disponibili. Questa malattia si caratterizza per la presenza di multipli schwannomi senza altre patologie associate.
Gli schwannomi possono essere singoli ed isolati oppure multipli e raggruppati, solitamente associati a sintomatologia dolorosa (dovuta all’effetto compressivo che esercitano le masse tumorali sugli altri organi). Le lesioni possono presentarsi a qualsiasi età e, diversamente dalla NF2, non riguardano i nervi vestibolari.
Foto e immagini
iStock.com/Sinhyu
iStock.com/DoloresGiraldez
iStock.com/chiewr
Diagnosi
Il percorso diagnostico per le neurofibromatosi ha subito un’importante evoluzione grazie al recente consenso internazionale che ha integrato l’analisi genetica nei criteri clinici classici. La diagnosi si basa oggi su una combinazione di osservazione dei segni fisici, anamnesi familiare e test molecolari avanzati.
Diagnosi di NF1
I criteri diagnostici per la NF1 sono stati aggiornati per distinguere precocemente la malattia da condizioni simili (come la sindrome di Legius). La diagnosi è confermata se sono presenti almeno due dei seguenti segni:
- Sei o più macchie “caffè-latte” di dimensioni superiori a 5 mm (prima della pubertà) o 15 mm (dopo la pubertà).
- Lentigginosi (piccole macchie scure) nelle zone ascellari o inguinali.
- Due o più neurofibromi di qualsiasi tipo o un neurofibroma plessiforme (più esteso e profondo).
- Due o più noduli di Lisch (amartomi dell’iride) o anomalie vascolari della coroide.
- Glioma delle vie ottiche.
- Lesioni ossee specifiche, come la displasia dello sfenoide o l’assottigliamento della corticale delle ossa lunghe (tibia).
- Identificazione di una mutazione patogenetica del gene NF1 tramite test genetico.
- Un genitore affetto da NF1 in base ai criteri sopra descritti.
L’indagine strumentale di supporto include la risonanza magnetica (RM) dell’encefalo e del midollo spinale per individuare eventuali tumori interni o aree di iperintensità (comunemente note come “oggetti luminosi non identificati” o UBO), tipiche dell’età pediatrica.
Diagnosi di NF2 e Schwannomatosi
Le linee guida attuali tendono a raggruppare queste condizioni sotto l’ombrello delle “schwannomatosi”, distinguendole in base al gene coinvolto. La diagnosi di NF2 (o schwannomatosi correlata a NF2) è considerata certa in caso di:
- Schwannomi vestibolari bilaterali rilevati tramite RM.
- Identificazione della mutazione del gene NF2 nel sangue o nei tessuti tumorali.
- Storia familiare positiva associata a schwannomi unilaterali o altri tumori tipici (meningiomi, ependimomi).
Per la Schwannomatosi non-NF2, la diagnosi richiede la presenza di due o più schwannomi non vestibolari, la conferma istologica e l’assenza di criteri clinici per la NF2, spesso supportata dal test genetico per i geni SMARCB1 o LZTR1.
Prognosi e complicazioni
Le neurofibromatosi hanno un’espressività ampiamente variabile, allo stesso modo anche la prognosi cambia da un caso all’altro. Grazie ai nuovi protocolli di monitoraggio e alle terapie farmacologiche mirate, l’aspettativa e la qualità della vita dei pazienti sono significativamente migliorate negli ultimi anni.
Le complicazioni possono verificarsi a qualsiasi età e sono conseguenti soprattutto all’effetto massa dei neurofibromi interni: sono possibili ad esempio problemi respiratori conseguenti all’ostruzione delle vie aeree da parte degli schwannomi interni. Inoltre, possono verificarsi nella NF1 deficit cognitivi, disturbi dell’attenzione e problemi psichiatrici.
I pazienti con neurofibromatosi di tipo 1 hanno un rischio stimato tra l’8% e il 15% di sviluppare tumori maligni della guaina dei nervi periferici (MPNST) durante la vita. Per questo motivo, la comparsa di dolore persistente, rapida crescita di un nodulo o perdita di forza richiede una valutazione medica immediata per escludere una trasformazione maligna.
Cura
Attualmente la gestione delle neurofibromatosi non si limita più alla sola attesa vigile, ma si avvale di un approccio multidisciplinare che coinvolge genetisti, neurologi, oncologi, dermatologi e chirurghi. Gli obiettivi principali della terapia sono il controllo della crescita tumorale, la riduzione del dolore e la prevenzione delle disabilità funzionali.
Le opzioni terapeutiche comprendono:
- Terapie farmacologiche mirate: L’introduzione dei MEK-inibitori (come il selumetinib) ha rivoluzionato il trattamento della NF1. Questi farmaci sono indicati per i neurofibromi plessiformi inoperabili e sintomatici nei bambini, mostrandosi efficaci nel ridurre il volume della massa tumorale.
- Chirurgia e tecniche mini-invasive: La rimozione chirurgica è indicata per tumori che causano dolore, compressione degli organi o gravi inestetismi. I neurofibromi cutanei possono essere trattati con successo tramite laser a CO2 o chirurgia plastica per migliorare l’aspetto estetico e il comfort del paziente.
- Gestione dei gliomi ottici: La maggior parte di questi tumori viene monitorata nel tempo. Se la vista è compromessa, si ricorre alla chemioterapia. La radioterapia è generalmente evitata nella NF1 a causa del rischio di indurre nuovi tumori secondari.
- Trattamento della NF2: Oltre alla chirurgia per la rimozione di meningiomi o schwannomi che comprimono il tronco encefalico, si possono utilizzare farmaci biologici (come il bevacizumab) per rallentare la perdita dell’udito e ridurre la crescita degli schwannomi vestibolari. Nei casi di sordità profonda, l’impianto cocleare o l’impianto al tronco encefalico rappresentano opzioni valide per il recupero uditivo.
- Terapia del dolore: Soprattutto nella schwannomatosi, il dolore neuropatico è il sintomo principale e viene gestito con farmaci specifici per il sistema nervoso o, in casi selezionati, con la rimozione chirurgica della lesione scatenante.
Stile di vita e supporto
Sebbene la dieta non influisca direttamente sulla crescita dei neurofibromi, uno stile di vita sano è fondamentale per gestire le comorbidità associate. Un’attività fisica regolare aiuta a contrastare l’ipotonia muscolare e i problemi di coordinazione spesso presenti nella NF1. È inoltre cruciale il supporto psicologico: convivere con una malattia cronica e potenzialmente visibile richiede un affiancamento costante per sostenere l’autostima e la salute mentale, specialmente durante l’adolescenza.
Il monitoraggio periodico resta il pilastro fondamentale: visite oculistiche regolari, controlli della pressione arteriosa e della crescita ossea nei bambini, insieme a una risonanza magnetica annuale o biennale (secondo il parere dello specialista), permettono di intervenire tempestivamente prima che insorgano complicazioni irreversibili.
Fonti e bibliografia
- Korf BR. Neurofibromatosis. Handb Clin Neurol. 2013;111:333-40. doi: 10.1016/B978-0-444-52891-9.00039-7.
- Neurofibromatosi di tipo 1
- Cainelli T., Giannetti A., Rebora A. Manuale di dermatologia medica e chirurgica. McGraw-Hill 4° edizione.
Tutti gli aggiornamenti su salute, alimentazione e benessere.






