Introduzione
Il retinoblastoma è un raro tipo di tumore agli occhi che di solito si sviluppa nella prima infanzia, in genere prima dei 5 anni. Questa forma di cancro colpisce la retina, il tessuto specializzato sensibile alla luce che si trova nella parte posteriore dell’occhio per rilevare luce e colore.
Nella maggior parte dei pazienti il tumore colpisce un solo occhio, ma un bambino su 3 mostra un interessamento di entrambi.
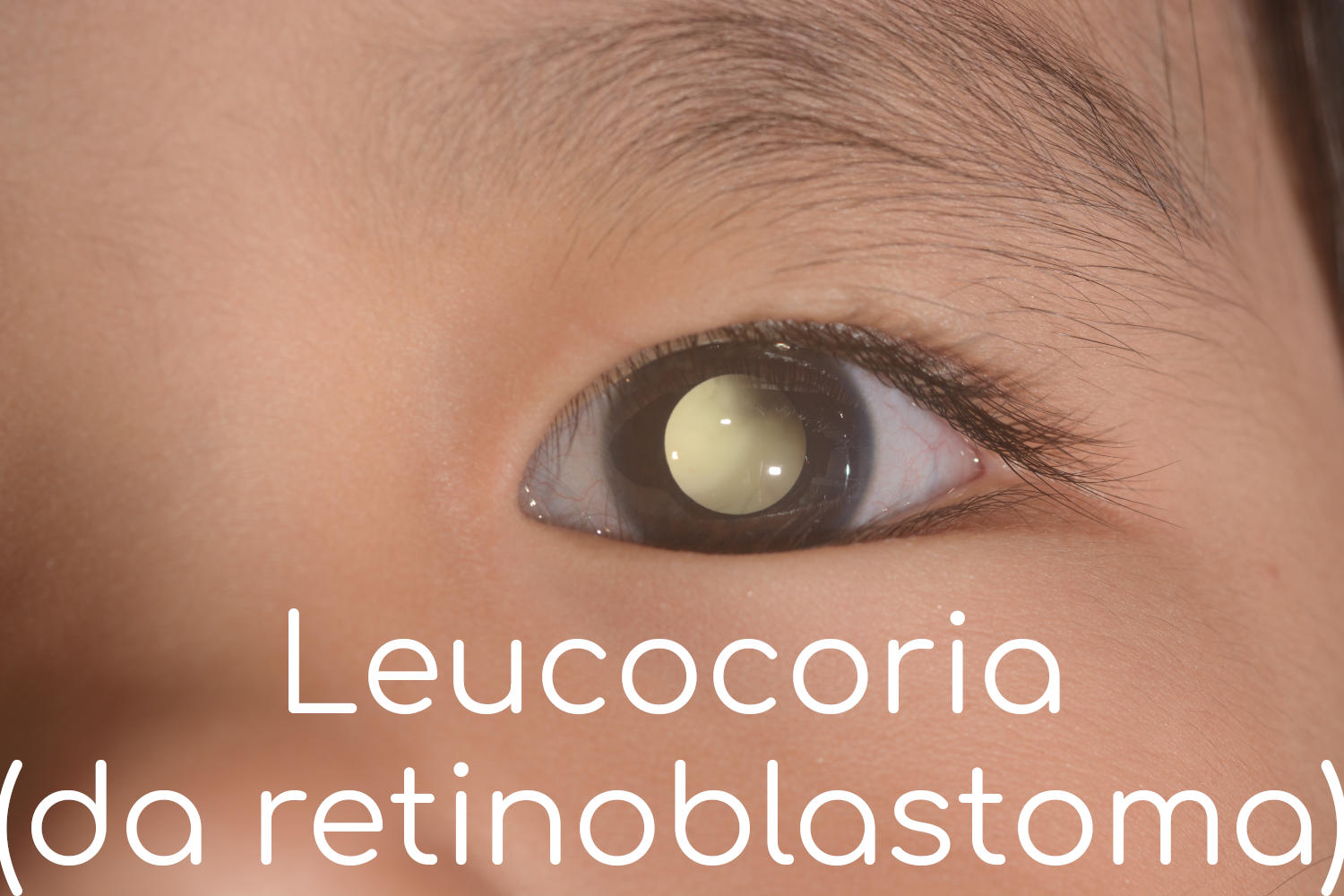
Il primo sintomo suggestivo di retinoblastoma è in genere un riflesso bianco visibile nella pupilla (leucocoria), che viene in genere notato in condizioni di scarsa illuminazione o nelle fotografie scattate con un flash. Altri segni e sintomi del retinoblastoma includono
- strabismo
- un cambiamento nel colore della parte colorata dell’occhio (iride);
- arrossamento, indolenzimento o gonfiore delle palpebre;
- riduzione della vista, fino alla cecità.
Sopravvivenza
Il retinoblastoma è spesso curabile quando viene diagnosticato precocemente tuttavia, se non viene diagnosticato e trattato tempestivamente, può diffondersi ad altre parti del corpo (metastasi) e diventare così pericoloso per la vita. Nei centri di assistenza specializzati, i tassi di sopravvivenza raggiungono il 95% circa, salvando anche la vista nella maggior parte dei casi (nei Paesi in via di sviluppo purtroppo le statistiche sono meno buone).
Cause
Il retinoblastoma è un tumore maligno raro che interessa circa un bambino su 18000 (rappresenta circa il 3% dei casi di tutti i tumori infantili).
L’età d’insorgenza è in genere entro il primo anno di vita in caso di malattia ad entrambi gli occhi, entro i 3 anni di età in caso di tumore in un solo occhio; è invece estremamente rara la comparsa dopo i 5 anni.
Nel complesso è possibile distinguere i seguenti casi:
- Circa il 25% dei pazienti sviluppa una forma bilaterale (entrambi gli occhi), che è sempre ereditaria,
- il 15% dei pazienti ha una forma ereditaria, ma limitata ad un solo occhio,
- il rimanente 60% presenta infine una forma monolaterale non ereditaria.
L’ereditarietà è principalmente autosomica dominante, ma con penetranza incompleta:
- autosomica: che colpisce porzioni del codice genetico non legate al sesso del paziente,
- dominante: è sufficiente ricevere un’unica copia difettosa del gene difettoso (ovvero da un solo genitore),
- penetranza incompleta: la sola presenza del gene difettoso NON implica necessariamente lo sviluppo del tumore.
Forma ereditabile
In questo tipo di retinoblastoma si rileva una mutazione del gene RB1 in tutte le cellule del corpo, ma da solo non è sufficiente ad indurre la comparsa di tumore.
I pazienti affetti sono tuttavia a rischio di diversi tumori non necessariamente oculari, come il pineoblastoma, l’osteosarcoma, i sarcomi dei tessuti molli e i melanomi (la possibilità di sviluppare un secondo tumore è del 6%, ma il rischio aumenta di cinque volte qualora si utilizzi la radioterapia per il trattamento del tumore primario).
Forma sporadica
I retinoblastomi non ereditabili interessano un unico occhio e non possono essere trasmessi ai figli; non vi è nemmeno un rischio aumentato dello sviluppo di tumori non oculari.
Nel caso di retinoblastoma in un solo occhio senza storia familiare positiva (ovvero senza parenti che ne siano stati affetti), si tratta presumibilmente di retinoblastoma non ereditabile e il rischio che si presenti anche in un fratello/sorella o figlio è stimato in 1 su 100 circa. in ciascun fratello e figlio del paziente è dell’1%.
Quasi il 90% dei casi di retinoblastomi unilaterali sono di forma non ereditaria.
Sintomi
Shutterstock/ARZTSAMUI
Il sintomo più comune del retinoblastoma è la leucocoria, ovvero una sorta di riflesso bianco nella pupilla (la parte nera dell’occhio):
- normalmente, quando viene colpita dalla luce, la pupilla appare rossa a causa dei vasi sanguigni presenti,
- mentre in caso di retinoblastoma si presenta bianca.
Soprattutto in passato in molti casi il riflesso veniva notato dai genitori in caso di foto scattate con il flash.
Tra gli altri possibili sintomi si segnalano:
- strabismo
- cambio del colore dell’iride
- occhi rossi ed infiammati (soprattutto in caso di sviluppo di glaucoma)
- lacrimazione
- visione ridotta
- limitazione dei movimenti oculari (più comune in caso di coinvolgimento di entrambi gli occhi).
Complicazioni
L’invasione del nervo ottico può verificarsi con la diffusione del tumore nello spazio subaracnoideo e nel cervello. La diffusione metastatica si verifica nei linfonodi regionali, nel fegato, nei polmoni, nelle ossa e nel cervello.
Il retinoblastoma può causare cecità se il trattamento non è sufficientemente tempestivo.
I bambini affetti dalla forma ereditaria di retinoblastoma corrono inoltre un rischio maggiore di sviluppare altri tipi di cancro negli anni successivi al trattamento e per questa ragione verranno pianificati periodici controlli.
Quando contattare il medico
Si raccomanda di chiedere il parere del pediatra in caso di:
- macchia bianca nell’occhio,
- disturbi di vista (che potrebbero emergere a seguito di ripetute cadute, necessità di avvicinare agli occhi i giochi, difficoltà a muoversi per casa).
Diagnosi
Il medico può evidenziare la presenza del riflesso bianco nell’occhio mediante un semplice esame dell’occhio (attraverso l’uso dell’oftalmoscopio).
In caso di rilievi suggestivi di anomalie è possibile ricorrere ad esami strumentali:
- ecografia (la sonda viene applicata sulla palpebra, ad occhio chiuso),
- risonanza magnetica.
Cura
L’approccio raccomandato per il retinoblastoma dipende essenzialmente dallo stadio del tumore al momento della diagnosi:
- intraoculare, il cancro è limitato all’interno del bulbo oculare,
- extraoculare, il cancro si è diffuso al tessuto circostante o, peggio, a distanza.
Quando il tumore si è diffuso diventa più difficile da curare, ma in Italia è una situazione ragionevolmente rara perché la condizione viene solitamente identificata ben prima che questo avvenga.
Le opzioni di trattamento per i tumori intraoculari sono il laser (fotocoagulazione) e il congelamento del tumore (crioterapia), somministrati con anestesia generale: in entrambi i casi l’obiettivo è quello di distruggere il tumore salvaguardando la vista.
In alcuni casi potrebbe essere necessaria la chemioterapia prima o dopo questi trattamenti.
I tumori più grandi richiedono approcci differenti (talvolta in combinazione):
- brachiterapia: se il tumore non è troppo grande, piccole placche radioattive vengono cucite sopra la massa tumorale e lasciate in posizione per alcuni giorni per distruggerlo;
- chemioterapia: può essere utilizzata per ridurre la dimensione del tumore all’inizio del trattamento, oppure può essere utile se esiste la possibilità che il cancro si diffonda (può essere tradizionale, via endovena, o somministrata direttamente nell’occhio);
- intervento chirurgico per rimuovere l’occhio: molto più usata in passato, ad oggi vi si ricorre quando non ci sia speranza di salvare la vista in altro modo.
Nei bambini trattati per il retinoblastoma esiste il rischio di recidiva, per questa ragione saranno necessari periodici controlli.
Fonti e bibliografia
- Retinoblastoma – Husnain Ishaq; Bhupendra C. Patel
- MedLinePlus
- AIRC
- NHS
- MSD
Tutti gli aggiornamenti su salute, alimentazione e benessere.