Introduzione
La sindrome del QT lungo (LQTS dall’inglese) è disturbo che colpisce l’attività elettrica del cuore.
Chi ne soffre può sviluppare un’aritmia in risposta all’esercizio fisico o allo stress, cioè un’anomalia del battito cardiaco che diventa irregolare e incontrollabile.
L’aritmia talvolta insorge senza cause apparenti, ma fortunatamente non tutti i soggetti interessati dalla sindrome soffrono anche di forme di aritmia pericolose, che tuttavia quando si verificano possono essere fatali.
L’espressione “QT lungo” si riferisce a una conformazione anomala evidenziata dall’ECG (elettrocardiogramma), l’esame che monitora l’attività elettrica del cuore.
Il QT è l’intervallo di tempo, registrato dall’elettrocardiogramma, che trascorre tra il momento in cui i ventricoli ricevono l’impulso a contrarsi e il momento in cui accumulano sufficiente potenziale per contrarsi di nuovo. Ricordiamo che i ventricoli sono le due “camere inferiori” del cuore.
La regolazione dell’attività elettrica che genera il battito cardiaco è complessa e l’organismo la controlla con particolare attenzione: in condizioni normali l’intervallo QT dura circa un terzo del tempo totale del battito registrato dall’elettrocardiogramma, invece nei pazienti che soffrono della sindrome questo intervallo dura più del normale.
La scansione temporale del battito cardiaco risulta quindi alterata e si possono sviluppare anomalie del ritmo cardiaco, in alcuni casi molto pericolose.
In molti pazienti la sindrome non è causa di sintomi e la diagnosi viene posta a seguito di un elettrocardiogramma (ECG) richiesto per altre ragioni; quando manifesta, la sindrome può causare la comparsa di:
- svenimento (il cuore smette di pompare sangue in modo regolare e il cervello, temporaneamente carente di ossigeno, risponde con lo sviluppo di sincope; in genere il ritmo del cuore ritorna normale entro un minuto o due e il paziente riprende conoscenza),
- convulsioni,
- palpitazioni cardiache.
Questi sintomi possono esordire in modo improvviso ed inaspettato, ma tra i fattori d’innesco più comuni ricordiamo:
- stress
- un rumore forte ed improvviso,
- sforzo fisico (per esempio sportivo),
- bradicardia durante il sonno (battito cardiaco rallentato).
La sindrome del QT lungo è comunque una malattia rara, ma studi recenti indicano una prevalenza di circa un caso ogni 2.500 persone, rendendola meno rara di quanto ipotizzato in passato.
Nei soli Stati Uniti la sindrome del QT lungo causa da 3.000 a 4.000 morti improvvise di bambini e giovani; la morte improvvisa senza motivi apparenti tra i bambini è un evento raro ma, se si verifica, spesso è causata da questa sindrome.
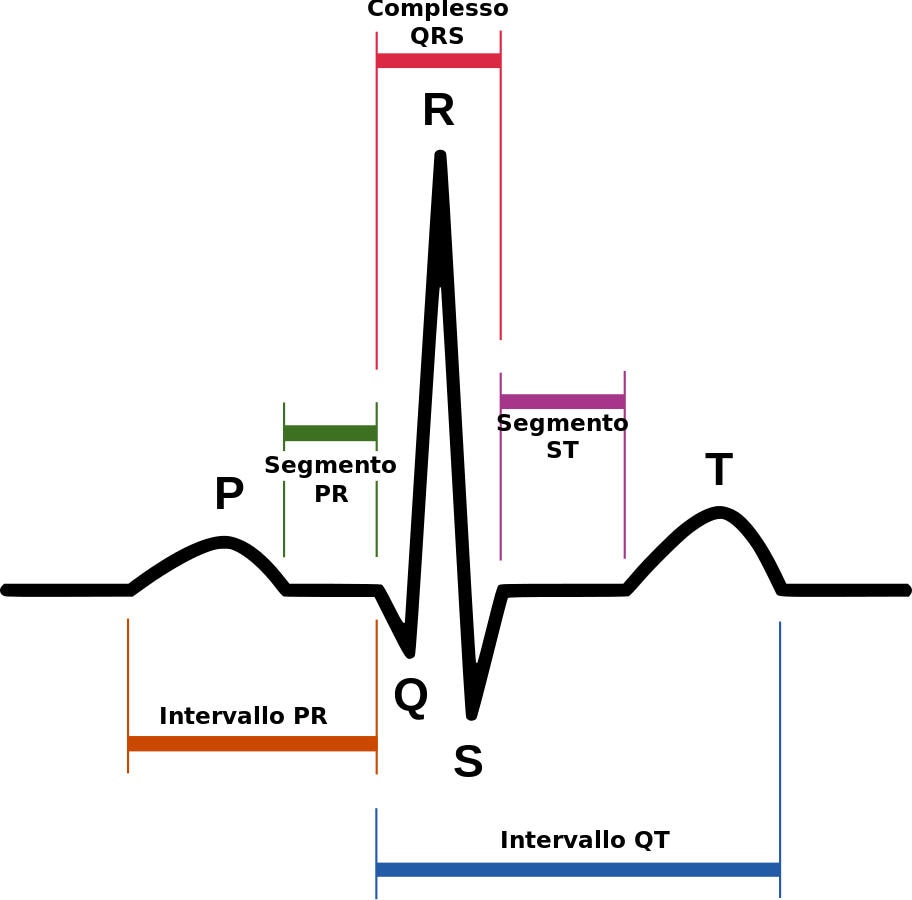
Di File:SinusRhythmLabels.svg: Created by Agateller (Anthony Atkielski), converted to svg by atom.opera derivata Cryptex – File:SinusRhythmLabels.svg, Copyrighted free use, Collegamento
Anatomia
Sulla superficie delle cellule del muscolo cardiaco si trovano minuscoli pori, chiamati canali ionici. I canali ionici si aprono e si chiudono permettendo agli ioni elettricamente carichi (sodio, calcio e potassio) di entrare e di uscire dalle cellule: in questo modo viene gestita l’attività elettrica del cuore.
L’attività elettrica permette quindi la contrazione del muscolo cardiaco: in condizioni normali l’impulso elettrico si trasmette da una cellula a quelle contigue in modo ordinato e coordinato, permettendo al cuore di pompare il sangue all’intero organismo.
Durante un battito normale le cellule muscolari delle camere superiori del cuore (gli atri) si contraggono, facendo fluire il sangue dagli atri verso i ventricoli; poi si contraggono le cellule muscolari dei ventricoli e quindi il sangue viene pompato verso i polmoni e verso il resto dell’organismo. Il processo coordinato di contrazione degli atri e dei ventricoli rappresenta un battito cardiaco normale.
Nei pazienti affetti da LQTS i problemi dei canali ionici delle cellule cardiache possono provocare anomalie dell’attività elettrica dei ventricoli: i canali ionici possono non funzionare correttamente, oppure possono essere in numero insufficiente, e in questa situazione il ritmo del cuore aumenta o presenta anomalie e quindi la vita del paziente può essere in pericolo.
Cause
LQTS ereditaria
La sindrome del QT lungo ereditaria è provocata dalla presenza di geni difettosi, in particolari quelli che controllano la produzione di determinati tipi di canali ionici del cuore:
- in alcuni casi i canali ionici possono essere in quantità insufficiente,
- oppure non funzionare correttamente,
- oppure ancora le due eventualità possono presentarsi contemporaneamente.
Se soffrite di LQTS ereditaria avete ricevuto il gene difettoso (o i geni difettosi) da uno dei genitori o da entrambi e in questo caso si tratta di un disturbo con cui dovrete convivere per tutta la vita.
Sono stati identificati molti tipi di LQTS ereditaria, ma i tipi più comuni rimangono l’1, il 2 e il 3.
- Se soffrite di LQTS 1 o di LQTS 2 il flusso degli ioni potassio attraverso i canali ionici delle cellule cardiache è anormale, e questo può causare problemi durante l’esercizio fisico (tipico della LQTS 1) oppure quando provate emozioni intense o venite spaventati da rumori improvvisi (tipico della LQTS 2). In queste situazioni può accadere che il cuore inizi a battere troppo velocemente.
- Se soffrite di LQTS 3 significa invece che c’è un’anomalia del flusso degli ioni sodio. In questo caso le aritmie tendono a verificarsi a riposo o durante il sonno, quando la frequenza cardiaca è fisiologicamente più lenta.
LQTS acquisita
Alcuni farmaci e alcune malattie possono provocare la sindrome del QT lungo acquisita (anche definta non ereditaria).
LQTS indotta dai farmaci
Attualmente un numero elevato di farmaci si sono dimostrati in grado di causare la LQTS, tra quelli di uso più frequente ricordiamo:
- antistaminici e decongestionanti,
- diuretici (farmaci che aiutano l’organismo a rimuovere l’acqua in eccesso),
- antibiotici (come l’eritromicina o la claritromicina),
- antidepressivi e antipsicotici,
- alcuni farmaci antiaritmici.
Alcuni dei pazienti che soffrono di sindrome del QT lungo indotta da farmaci potrebbero anche soffrire di una forma ereditaria latente del disturbo, che si manifesta solo in presenza di queste sostanze o di squilibri elettrolitici.
Altre cause dell’LQTS
La diarrea o il vomito protratti, che impoveriscono il sangue degli ioni potassio o sodio (ipopotassiemia o iponatriemia), possono causare il prolungamento del QT. La condizione rientra generalmente con la correzione dei livelli elettrolitici.
Anche l’anoressia nervosa e la bulimia, così come alcuni disturbi della tiroide, rappresentano fattori di rischio significativi per le alterazioni elettrolitiche che scatenano la LQTS.
Fattori di rischio
La LQTS ereditaria di solito è diagnosticata per la prima volta durante l’infanzia o in età giovanile. Il rischio di eventi aritmici gravi è maggiore nei maschi prima della pubertà e nelle femmine dopo la pubertà.
Le donne presentano un rischio aumentato durante il ciclo mestruale e, in particolare, nel periodo del post-partum.
Esistono inoltre rare varianti (come la sindrome di Jervell e Lange-Nielsen) che associano il QT lungo alla sordità congenita.
Sintomi
Se soffrite di sindrome del QT lungo siete potenzialmente soggetti ad un’aritmia improvvisa e pericolosa, come la tachicardia ventricolare a “torsione di punta”, che può degenerare in fibrillazione ventricolare.
I segni e i sintomi compaiono spesso durante l’infanzia o l’adolescenza e comprendono:
- Svenimenti (sincope) improvvisi, spesso innescati da stress emotivo, fisico o stimoli uditivi.
- Convulsioni che possono essere erroneamente diagnosticate come epilessia, ma che sono in realtà dovute alla temporanea mancanza di ossigeno al cervello durante l’aritmia.
- Annegamento improvviso o incidenti inspiegabili durante il nuoto.
- Arresto cardiaco improvviso, che rappresenta purtroppo il sintomo d’esordio in una piccola percentuale di casi.
LQTS asintomatica
Molte persone sono portatrici della sindrome senza saperlo perché non manifestano alcun sintomo (LQTS silente). In questi casi, la diagnosi avviene spesso durante screening familiari o esami eseguiti per altri motivi.
Diagnosi
La diagnosi della sindrome del QT lungo richiede una valutazione specialistica cardiologica approfondita. Il percorso diagnostico moderno non si limita alla semplice misurazione di un valore sull’elettrocardiogramma, ma integra criteri clinici, strumentali e genetici per definire il profilo di rischio del paziente.
Valutazione clinica e Score di Schwartz
Il punto di partenza è l’anamnesi personale e familiare. Il medico indagherà la presenza di episodi passati di svenimento (specialmente se legati a stress o sforzo), convulsioni non spiegate o casi di morte improvvisa in famiglia in giovane età (sotto i 40-50 anni).
Per standardizzare la diagnosi, i cardiologi utilizzano spesso lo Score di Schwartz, un sistema a punteggio che assegna valori diversi in base a:
- Parametri elettrocardiografici (durata del QT corretto, morfologia dell’onda T, presenza di alternanza dell’onda T).
- Storia clinica (sincopi, sordità congenita).
- Anamnesi familiare (membri della famiglia con LQTS accertata o morte improvvisa).
Elettrocardiogramma (ECG) e monitoraggio
L’elettrocardiogramma a riposo è l’esame fondamentale. Il medico calcola l’intervallo QT corretto (QTc) utilizzando formule matematiche (come la formula di Bazett) per compensare l’effetto della frequenza cardiaca. Poiché l’intervallo QT può variare durante la giornata, un singolo ECG normale non esclude la diagnosi.
Per una valutazione più completa possono essere necessari:
- Monitoraggio Holter (24-48 ore): Registra l’attività elettrica durante le normali attività quotidiane, permettendo di osservare come il QT si adatta alle variazioni fisiologiche del battito o durante il sonno.
- Test da sforzo: Fondamentale per evidenziare un mancato accorciamento del QT durante l’esercizio o anomalie nella fase di recupero, tipiche della variante LQTS 1.
- Test di provocazione: In casi dubbi e in ambiente protetto, possono essere somministrati farmaci (come l’adrenalina) per osservare la reazione del sistema elettrico cardiaco.
Test genetici e screening familiare
L’analisi del DNA è oggi parte integrante del protocollo diagnostico. Identificare la mutazione specifica permette di:
- Confermare la diagnosi nei casi clinici dubbi.
- Identificare il sottotipo di sindrome (LQT1, LQT2, LQT3, ecc.), dato essenziale per personalizzare la terapia.
- Eseguire uno screening mirato sui familiari (“screening a cascata”), permettendo di identificare soggetti a rischio anche se asintomatici o con ECG normale.
Sebbene i test genetici siano altamente avanzati, circa il 20-25% dei pazienti con diagnosi clinica certa non presenta una mutazione nei geni attualmente noti; un test genetico negativo, quindi, non esclude necessariamente la malattia se i segni clinici sono evidenti.
Cura e terapia
Gli obiettivi principali della cura sono la prevenzione delle aritmie ventricolari maligne, la riduzione del rischio di sincope e la protezione dalla morte improvvisa. La gestione della sindrome del QT lungo è altamente personalizzata e si basa sul genotipo, sul sesso e sulla storia clinica del paziente.
Terapia farmacologica
I farmaci rappresentano la prima linea di trattamento per la maggior parte dei pazienti. Non “accorciano” necessariamente il QT in modo visibile sull’ECG, ma agiscono come uno scudo contro gli stimoli che scatenano le aritmie.
- Betabloccanti: Sono i farmaci di scelta. Agiscono riducendo la risposta del cuore all’adrenalina e allo stress. Le linee guida attuali privilegiano l’uso di molecole come il nadololo o il propranololo, che si sono dimostrate più efficaci nel prevenire eventi cardiaci rispetto ad altri betabloccanti di vecchia generazione. Sono particolarmente efficaci nei pazienti con LQT1 e LQT2.
- Bloccanti dei canali del sodio: Per i pazienti con LQT3, la terapia farmacologica può includere farmaci come la mexiletina. Questa molecola agisce specificamente sull’anomalia del canale del sodio, contribuendo ad accorciare l’intervallo QT e a stabilizzare il ritmo cardiaco, specialmente durante il riposo.
Denervazione simpatica cardiaca sinistra (LCSD)
Si tratta di un intervento chirurgico mini-invasivo che consiste nella rimozione di alcuni nervi del sistema simpatico che raggiungono il cuore. È un’opzione terapeutica consolidata per i pazienti che:
- Continuano ad avere sintomi nonostante la terapia farmacologica ottimale.
- Non tollerano gli effetti collaterali dei betabloccanti.
- Presentano un rischio elevato ma non desiderano o non possono impiantare un defibrillatore.
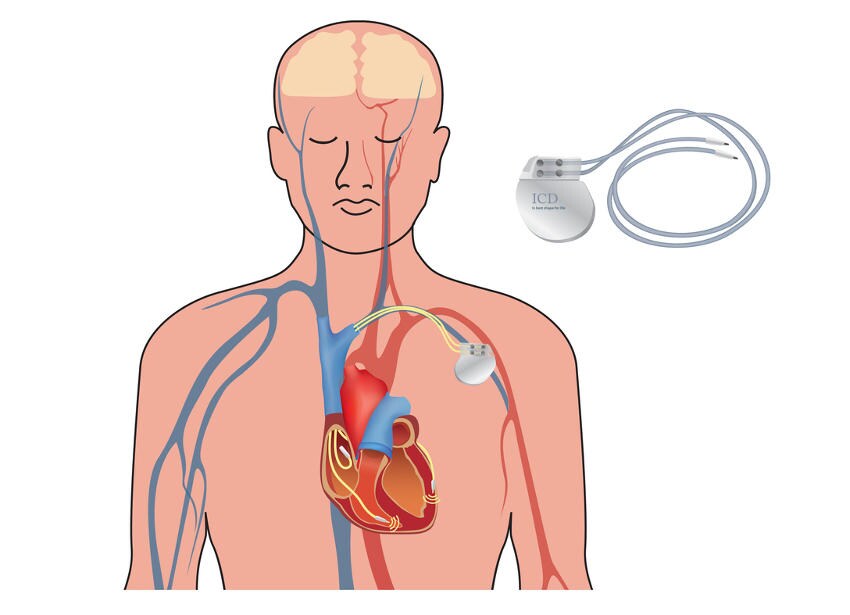
Defibrillatore automatico impiantabile (ICD)
L’ICD è un dispositivo elettronico che monitora costantemente il ritmo cardiaco e interviene con una scarica elettrica se rileva un’aritmia fatale. L’impianto è raccomandato per i pazienti ad altissimo rischio, in particolare per coloro che sono già sopravvissuti a un arresto cardiaco o che continuano a presentare sincopi gravi nonostante la terapia con betabloccanti e/o la chirurgia.
Stile di vita e precauzioni
Il coinvolgimento attivo del paziente è fondamentale per il successo della terapia. Le raccomandazioni includono:
- Evitare farmaci a rischio: È fondamentale non assumere farmaci che prolungano l’intervallo QT. I pazienti dovrebbero sempre consultare elenchi aggiornati (come quelli disponibili su portali specialistici certificati) e informare ogni medico o dentista della propria condizione.
- Gestione degli elettroliti: Mantenere livelli adeguati di potassio e magnesio è cruciale. Situazioni che causano perdita di liquidi, come febbre alta, diarrea o vomito, devono essere gestite con tempestività per evitare squilibri che favoriscono le aritmie.
- Attività fisica: Le restrizioni variano a seconda del tipo di QT lungo. Mentre un tempo si vietava lo sport a tutti, oggi l’approccio è più flessibile: molti pazienti possono praticare attività fisica moderata sotto stretto controllo medico, sebbene gli sport agonistici ad alto impatto restino sconsigliati per i profili di rischio più elevati.
- Sicurezza ambientale: Per i pazienti con LQT2, è prudente ridurre gli stimoli sonori improvvisi (sveglie a volume moderato, rimozione di telefoni dalla camera da letto).
La prognosi per i pazienti con sindrome del QT lungo è oggi eccellente nella stragrande maggioranza dei casi, a condizione che la diagnosi sia precoce e la terapia venga seguita con rigorosa aderenza.
Fonte principale
Le domande più frequenti
Cos'è l'intervallo QT?
Cosa significa QTc?
Come si calcola il QTc?
Quando preoccuparsi per la sindrome del QT lungo?
Quali sono i segnali di allerta che richiedono un ECG o una visita cardiologica?
Il QT lungo può essere causato dai farmaci?
È sicuro continuare a praticare sport se si ha il QT lungo?
Tutti gli aggiornamenti su salute, alimentazione e benessere.






{kind=link}
{kind=link}