Introduzione
La Sindrome di Brugada è una malattia genetica caratterizzata da disturbi dell’attività elettrica del cuore, in assenza di evidenti alterazioni strutturali a carico del miocardio.
La Sindrome di Brugada fu descritta per la prima volta da autori italiani nel 1988, nonostante i reperti elettrocardiografici caratteristici della malattia fossero già evidenti nel 1953 ad Osher e Wolff, i quali rilevarono anomalie dinamiche all’ECG che simulavano un infarto del miocardio in un soggetto maschio sano.
Questa sindrome oggi è conosciuta con riferimento al nome dei fratelli Brugada, i quali la descrissero in maniera dettagliata nel 1992.
Alcuni pazienti ereditano il disturbo dai genitori mentre altri, invece, lo sviluppano tra i 30 e i 40 anni in modo del tutto inspiegabile.
I pazienti che ne sono affetti presentano una predisposizione maggiore a sviluppare aritmie ventricolari maligne e sono considerati a rischio di morte improvvisa.
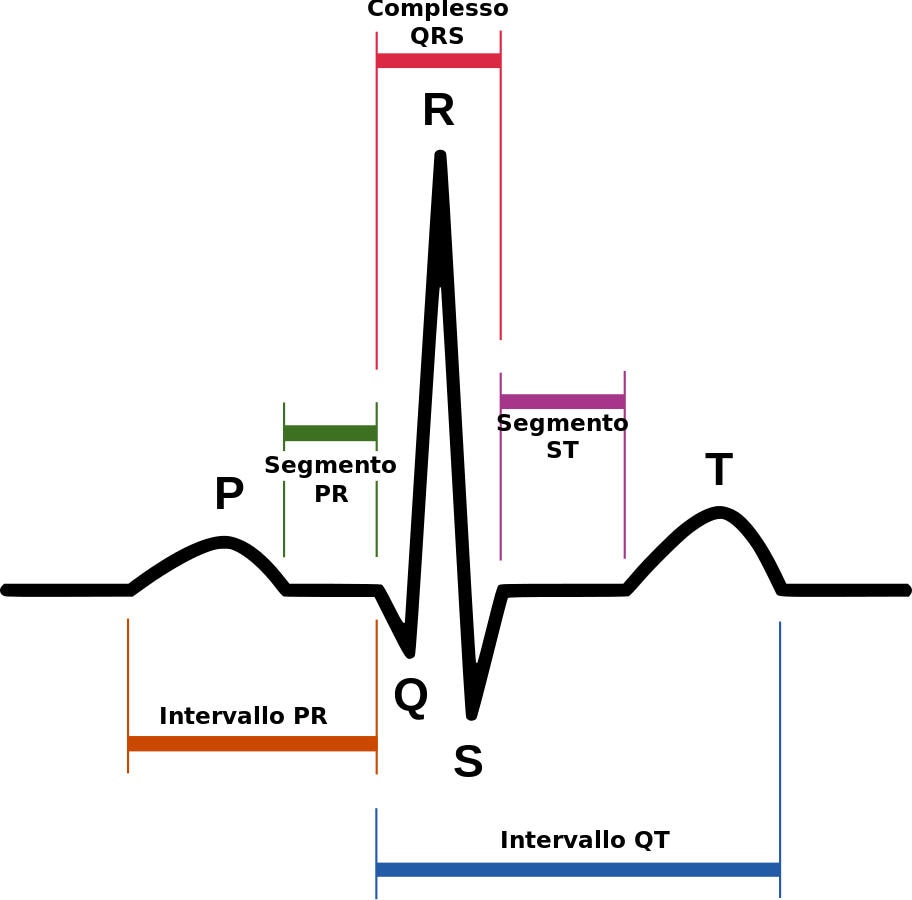
Nella maggior parte dei casi, la diagnosi è posta tramite il rilievo di alterazioni caratteristiche visibili all’elettrocardiogramma (quali un blocco di branca destro e un sopraslivellamento del tratto ST nelle derivazioni precordiali destre). Questi reperti elettrocardiografici possono
- variare nel tempo,
- presentarsi in maniera incostante
- o essere accentuati
- dall’assunzione di farmaci (principalmente bloccanti i canali del sodio, beta-bloccanti, litio, antidepressivi triciclici)
- o da alcune condizioni fisiche (come ad esempio la febbre).
Poiché la malattia può essere familiare, viene raccomandato uno screening dei parenti dei pazienti affetti, per valutare il rischio di malattia.
In alcuni pazienti la sindrome di Brugada è asintomatica, tuttavia, in molti casi, le aritmie ventricolari possono generare specialmente di notte e senza correlazione con l’esercizio fisico
- palpitazioni,
- sincope,
- aritmie ventricolari,
- arresto cardiaco (nei casi più gravi).
Nei pazienti affetti da Sindrome di Brugada si raccomanda di evitare l’assunzione di farmaci e l’esposizione ad eventi che possano favorire il manifestarsi di episodi aritmici; nei soggetti con pregresso arresto cardiaco e in quelli in cui uno studio elettrofisiologico abbia dimostrato la possibilità che si verifichino aritmie ventricolari maligne è raccomandato l’impianto di un defibrillatore automatico, in grado di controllare in continuo il ritmo cardiaco e di intervenire in caso si generino aritmie pericolose per la vita.
Poiché la Sindrome di Brugada non ha una progressione clinica definita, è possibile che il paziente presenti un basso rischio aritmico per tutta la vita, ma è comunque necessario monitorare frequentemente la malattia attraverso visite cardiologiche programmate ed eventualmente valutare altre opzioni terapeutiche, quali l’uso di farmaci antiaritmici, come ad esempio la chinidina.
Cosa sono le aritmie cardiache?
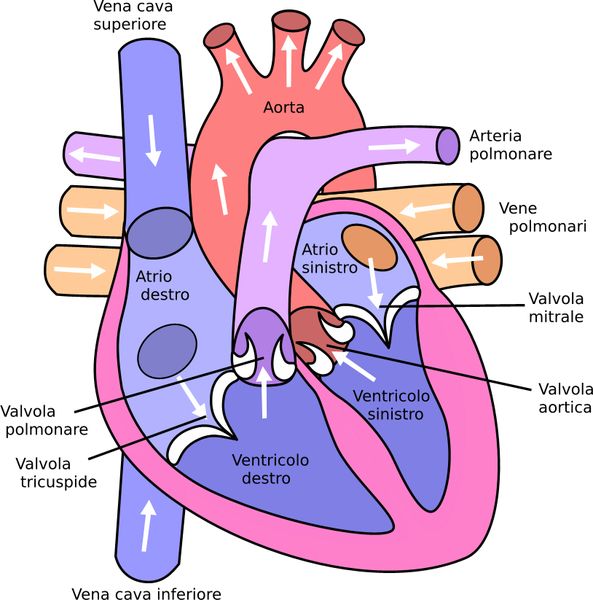
L’impulso elettrico alla base della conduzione cardiaca prende di norma origine da una struttura posta in corrispondenza dell’atrio destro, il nodo seno-atriale, il quale, fungendo da pace-maker naturale del cuore, rappresenta il segnapassi dominante che fa si che l’impulso si propaghi dagli atri fino a raggiungere i ventricoli, stimolandone la contrazione.
Questo processo consente all’attività cardiaca di svolgersi con un “ritmo sinusale”, che ha una frequenza compresa tra i 60 e i 100 battiti al minuto in condizioni di riposo.
Le alterazioni di questo processo sono principalmente tre ed è sufficiente una sola di queste condizioni perché insorga un’aritmia:
- Modificazioni della frequenza e della regolarità del battito cardiaco; parliamo di tachicardia quando il battito è accelerato (oltre 100 battiti per minuto) o di bradicardia quando è più lento (sotto i 60 battiti al minuto).
- Variazioni del segnapassi dominante, rappresentato principalmente dal nodo seno-atriale, in grado di garantire la “normalità” del battito cardiaco.
- Disturbi di conduzione e propagazione dell’impulso di contrazione con alterazioni del ritmo cardiaco.
Cause
La malattia colpisce prevalentemente le popolazioni di origine asiatica e risulta essere più frequente nei maschi di età compresa tra i 30 e il 40 anni, specialmente se con storia familiare alle proprie spalle.
Nella maggior parte dei casi alla base di questa sindrome c’è un’anomalia funzionale genetica trasmessa dai genitori ai figli con eredità autosomica dominante; per la restante parte dei malati, l’origine della malattia resta invece sconosciuta o non meglio chiarificata.
Le mutazioni genetiche, riguardano principalmente un gene situato sul cromosoma 3, SCN5A, che produce proteine simili a canali che, localizzandosi in corrispondenza della membrana delle cellule muscolari cardiache, consentono il passaggio degli ioni sodio, generando una vera e propria corrente che è necessaria per la trasmissione degli impulsi elettrici e quindi per la contrazione del cuore.
I pazienti con Sindrome di Brugada presentano un malfunzionamento di questi canali, che non possono garantire un battito cardiaco adeguato e possono altresì determinare squilibri nell’attività elettrica cardiaca, aumentando il rischio di insorgenza di aritmie potenzialmente fatali.
Sebbene la mutazione di SCN5A renda ragione del 18-30% dei casi di Sindrome di Brugada, con il progredire della ricerca sono state evidenziate altre mutazioni a carico di diversi geni, come:
- GPD1L : produce la glicerolo-3-fosfato deidrogenasi e pur non essendo un canale di membrana, può modulare gli scambi ionici che avvengono all’interno di essa;
- CACNA1 e CACNB2: producono canali di membrana per il calcio, il cui malfunzionamento può alterare la trasmissione dell’impulso contrattile nel cuore;
- SCN1B e SCN10A: due canali per il sodio, individuati nel 2013 ed ipoteticamente coinvolti nella genesi della malattia;
- HEY2 : fattore di trascrizione, scoperto nel 2013 e con un ruolo nella genesi della malattia ancora non ben chiarificato.
Per i casi di pazienti affetti in cui la Sindrome di Brugada non possa essere spiegata da una mutazione genetica sottostante, sono stati ipotizzati alcuni motivi alla base dell’incorretta trasmissione degli impulsi elettrici nel cuore, tra cui:
- uso di droghe, principalmente cocaina;
- ipertensione (pressione alta);
- angina pectoris;
- squilibri elettrolitici ( riguardanti la concentrazione di ioni calcio, sodio, potassio, nel sangue);
- assunzione di alcuni farmaci (principalmente bloccanti i canali del sodio, beta-bloccanti, litio, antidepressivi triciclici);
- stati febbrili.
Bisogna considerare inoltre maggiormente a rischio di malattia i pazienti che presentino una storia familiare di morte improvvisa in età inferiore ai 45 anni.
Sintomi
La malattia si presenta con una grande variabilità clinica e l’elettrocardiogramma del paziente affetto può essere soggetto a variazioni e apparire patologico o sostanzialmente normale, anche nell’ambito della stessa giornata.
I pazienti possono essere asintomatici o presentare:
- Episodi di sincope (svenimenti), spesso non preceduta da avvisaglie, sono provocati dalla contrazione rapida e non efficace dei ventricoli che solitamente torna normale dopo qualche secondo; poiché la perdita di coscienza è immediata, senza sintomi premonitori, i pazienti sono anche a rischio di traumi.
- Episodi di cardiopalmo ( sensazione soggettiva di palpitazione cardiaca), spesso associato a malessere.
- Episodi di enuresi notturna (“fare pipì a letto”), nel caso in cui la sincope aritmica si verifichi durante la notte con rilascio sfinteriale.
- Fibrillazione ventricolare documentata.
- Tachicardia ventricolare polimorfa.
- Respiro agonico notturno (“boccheggiamento “con riduzione della frequenza degli atti respiratori).
I sintomi si manifestano maggiormente a riposo, durante la notte o durante la fase di recupero dall’esercizio fisico, mentre l’attivazione adrenergica (in risposta a stress psichico o fisico) così come l’esercizio fisico, non costituiscono un fattore favorente le aritmie che si manifestano nella patologia.
Complicazioni
La morte cardiaca improvvisa è la manifestatione più tragica della malattia e di solito si verifica maggiormente nei soggetti tra i 25 e i 50 anni.
È dovuta a un arresto cardiaco determinato dalla fibrillazione ventricolare; in questo caso le camere inferiori del cuore, i ventricoli, si contraggono in maniera rapidissima e irregolare, generando una contrazione non valida che può far cessare l’attività cardiaca, determinando morte improvvisa.
Diagnosi
La diagnosi della Sindrome di Brugada rappresenta una sfida clinica a causa della natura transitoria e dinamica delle alterazioni elettriche. Il percorso diagnostico moderno non si limita alla semplice lettura di un tracciato, ma integra dati clinici, strumentali e genetici per definire il profilo di rischio del paziente.
Elettrocardiogramma (ECG) a riposo
Lo strumento fondamentale rimane l’elettrocardiogramma a 12 derivazioni. Per aumentare la sensibilità del test, i cardiologi utilizzano spesso il posizionamento dei “precursori alti”, spostando gli elettrodi precordiali (V1 e V2) dal quarto al secondo o terzo spazio intercostale. Si distinguono due pattern principali:
- Tipo 1 (diagnostico): caratterizzato da un sopraslivellamento del tratto ST a “forma di tenda” o “coved” (convesso verso l’alto) di almeno 2 mm, seguito da un’onda T negativa. Questo pattern, se spontaneo, è sufficiente per la diagnosi in presenza del quadro clinico appropriato.
- Tipo 2 (sospetto): presenta un sopraslivellamento dell’ST con morfologia “a sella” (saddle-back). Pur non essendo di per sé diagnostico, richiede ulteriori approfondimenti perché può convertirsi nel Tipo 1 in determinate circostanze.
Test provocativi ai bloccanti dei canali del sodio
Poiché molti pazienti presentano un ECG basale silente o non conclusivo, si ricorre ai test farmacologici. Durante l’esame, eseguito sotto stretto monitoraggio in ambiente ospedaliero, vengono somministrati farmaci come la flecainide o l’ajmalina. Questi principi attivi “smascherano” la sindrome inducendo la comparsa del pattern Tipo 1 nei soggetti predisposti. Un test positivo conferma la diagnosi, ma la sua interpretazione deve sempre essere contestualizzata dall’elettrofisiologo.
Monitoraggio Holter e Studio Elettrofisiologico (SEF)
L’elettrocardiogramma secondo Holter è essenziale per catturare eventuali aritmie notturne o fluttuazioni spontanee del tracciato durante le 24 o 48 ore. Lo Studio Elettrofisiologico (SEF), invece, è un esame invasivo che prevede l’introduzione di cateteri nel cuore per testare la vulnerabilità del miocardio a sviluppare aritmie ventricolari maligne. Attualmente, il SEF viene utilizzato soprattutto per la stratificazione del rischio nei pazienti asintomatici con pattern spontaneo di Tipo 1.
Analisi genetica e screening familiare
Il test genetico per la ricerca di mutazioni (principalmente a carico del gene SCN5A) è raccomandato dopo la diagnosi clinica del “caso indice”. È importante sottolineare che un test genetico negativo non esclude la malattia (poiché molte mutazioni restano ancora sconosciute), ma un risultato positivo permette uno screening mirato e precoce di tutti i familiari di primo grado, che dovrebbero comunque essere sottoposti a una valutazione cardiologica completa.
Diagnosi differenziale
Per una diagnosi accurata, il medico deve escludere altre condizioni che possono simulare il pattern di Brugada (fenocopie), come l’ecocardiogramma per escludere malattie strutturali del cuore, squilibri elettrolitici gravi, o l’uso di specifici farmaci psicotropi.
Cura
L’obiettivo primario della terapia nella Sindrome di Brugada è la prevenzione della morte improvvisa causata da fibrillazione ventricolare. Non esiste attualmente una cura definitiva che corregga l’anomalia genetica, pertanto la gestione si concentra sulla protezione del paziente e sulla prevenzione dei fattori scatenanti.
Misure comportamentali e stile di vita
Ogni paziente diagnosticato, anche se asintomatico, deve adottare precauzioni rigorose per ridurre il rischio aritmico:
- Gestione della febbre: la temperatura elevata è uno dei principali trigger per le aritmie di Brugada. È fondamentale trattare tempestivamente ogni stato febbrile con antipiretici (come il paracetamolo) e spugnature fredde.
- Revisione della terapia farmacologica: il paziente deve consultare costantemente il portale di riferimento (BrugadaDrugs.org) ed evitare assolutamente farmaci controindicati, inclusi alcuni anestetici, antiaritmici e antidepressivi.
- Consumo di alcol e pasti abbondanti: è raccomandata cautela con l’alcol e i pasti eccessivamente pesanti (soprattutto serali), poiché possono aumentare il tono vagale e favorire l’insorgenza di aritmie notturne.
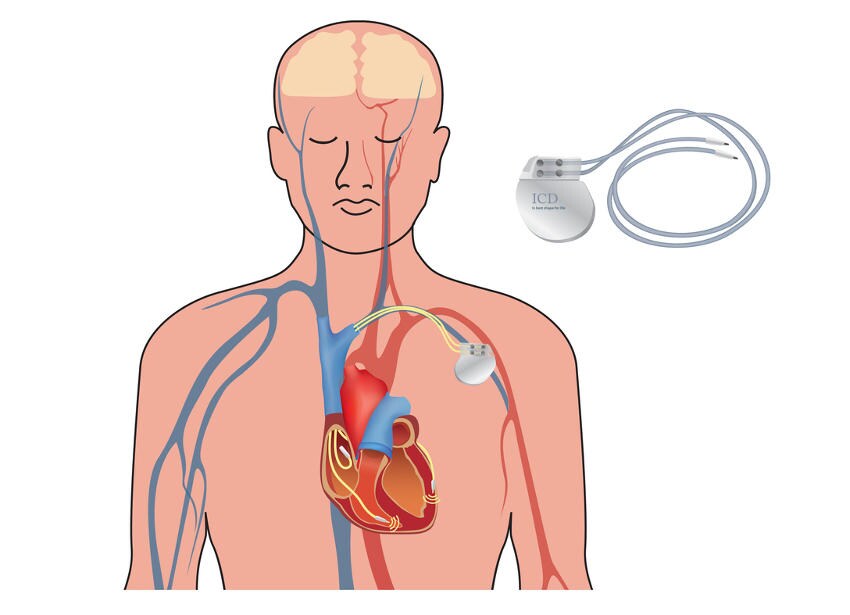
Defibrillatore Automatico Impiantabile (ICD)
L’impianto di un defibrillatore (ICD) rappresenta l’unica terapia salvavita di comprovata efficacia per i pazienti ad alto rischio (coloro che hanno già subito un arresto cardiaco o presentano sincope di sospetta origine aritmica).
iStock.com/Terriana
Il dispositivo monitora costantemente il ritmo cardiaco e interviene con una scarica elettrica per interrompere aritmie pericolose. Negli ultimi anni, in casi selezionati, si preferisce l’ICD sottocutaneo (S-ICD), che evita l’inserimento di cateteri all’interno dei vasi sanguigni e del cuore, riducendo alcune complicazioni a lungo termine.
Terapia farmacologica (Chinidina)
In casi specifici, quando l’ICD non è indicato o in presenza di frequenti interventi del defibrillatore (“tempeste elettriche”), può essere prescritta la chinidina. Questo farmaco agisce inibendo i canali del potassio e stabilizzando l’attività elettrica del cuore. Sebbene efficace nel normalizzare il tracciato ECG e ridurre le aritmie, può presentare effetti collaterali (specialmente gastrointestinali) che richiedono un attento monitoraggio medico.
Ablazione transcatetere
Una delle innovazioni più significative nel trattamento della sindrome è l’ablazione epicardica del tratto di efflusso del ventricolo destro (RVOT). Questa procedura mininvasiva mira a “isolare” o eliminare le aree del tessuto cardiaco responsabili delle anomalie elettriche. Viene proposta tipicamente a pazienti con episodi aritmici ricorrenti nonostante il defibrillatore o in centri di eccellenza come alternativa terapeutica in casi selezionati, mostrando risultati promettenti nella normalizzazione del pattern ECG e nella riduzione del carico aritmico.
Fonti e bibliografia
Tutti gli aggiornamenti su salute, alimentazione e benessere.