Introduzione
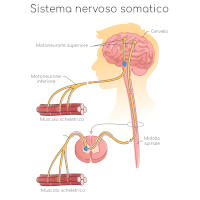
La malattia di Charcot-Marie-Tooth fa parte di un gruppo di disturbi causa di danni ai nervi periferici, ovvero quelli che trasmettono segnali dal sistema nervoso centrale (cervello e midollo spinale) al resto del corpo e quelli sensoriali (in senso inverso, in direzione del cervello)
Nota anche come neuropatia motorio-sensitiva ereditaria, è uno dei disturbi neurologici ereditari più comuni, interessando circa 2,6 milioni di persone in tutto il mondo; spesso indicata semplicemente con l’acronimo CMT, che deriva dal cognome dei tre medici che per primi la descrissero nel 1886, può purtroppo interferire con il funzionamento dei nervi responsabili del controllo muscolare volontario, causando una progressiva debolezza muscolare che diventa in genere evidente nell’adolescenza o nella prima età adulta (anche se l’insorgenza della malattia può comunque verificarsi a qualsiasi età). Poiché vengono colpiti per primi i nervi più lunghi, i sintomi di solito compaiono inizialmente nei piedi e nella parte inferiore delle gambe, cui seguono dita, mani e braccia.
Nel complesso la maggior parte dei pazienti manifesta quindi una certa disabilità fisica, anche se una minoranza potrebbe in realtà non diventare mai consapevole di esserne affetta.
Attualmente non esiste una cura per la CMT, che tuttavia può essere gestita con una terapia di supporto; la patologia in genere non diventa pericolosa per la vita e solo raramente colpisce i muscoli coinvolti in funzioni vitali come la respirazione, quindi i pazienti godono in genere di un’aspettativa di vita del tutto sovrapponibile a quella della popolazione generale.
A chi rivolgersi
Si segnala l’esistenza in Italia dell’associazione ACMT-Rete dedicata, tra l’altro, a “sviluppare l’incontro e il confronto fra le persone affette dalla malattia di Charcot-Marie-Tooth e i loro familiari”, che può essere di grande aiuto per tutti coloro si trovino a dover affrontare le sfide che la malattia impone.
Shutterstock/Kateryna Kon
Cause
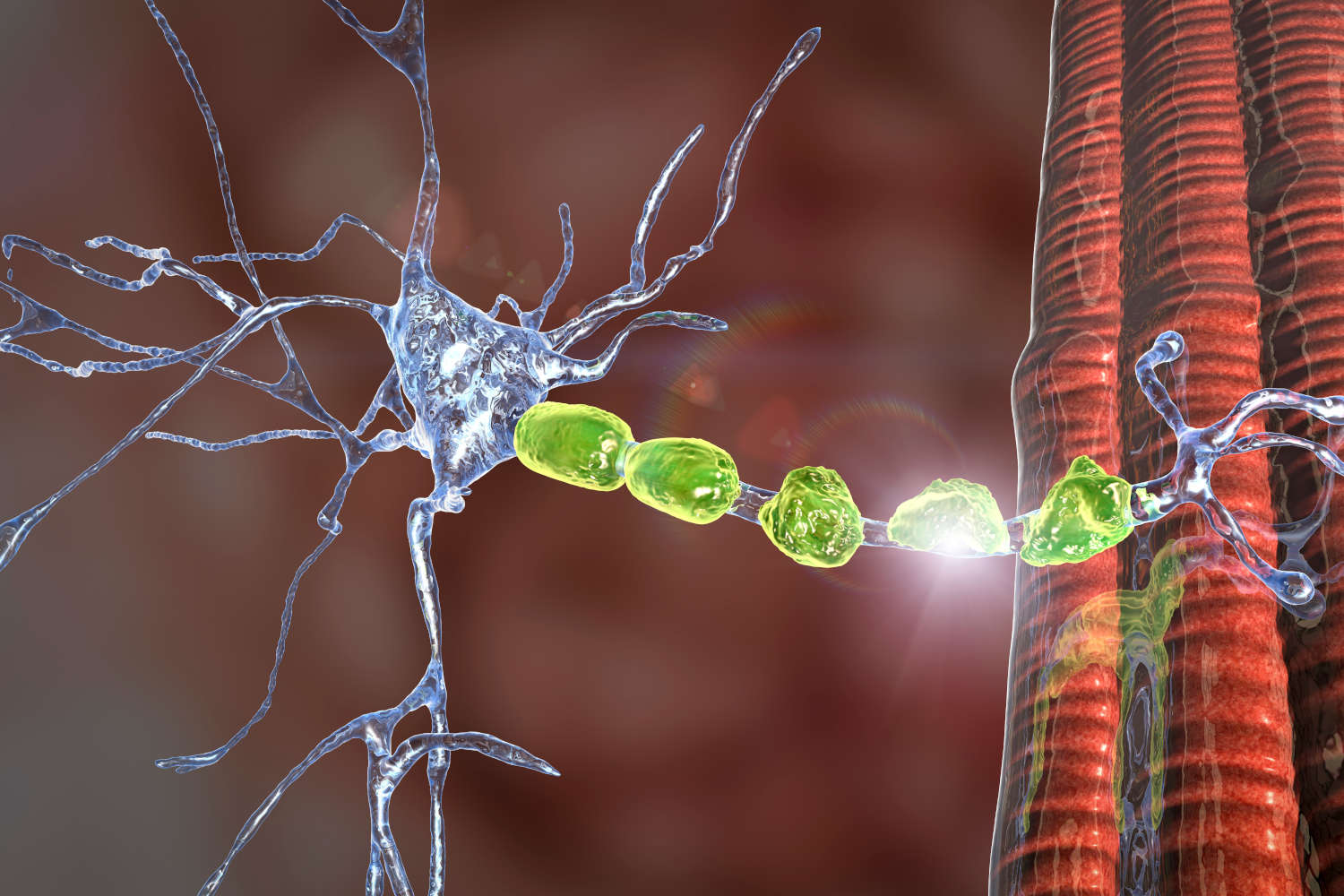
La trasmissione dei segnali nervosi da e verso il sistema nervoso centrale avviene attraverso l’invio di segnali elettrici lungo una lunga e sottile protuberanza della cellula nervosa, che prende il nome di assone. Come un cavo elettrico, l’assone è circondato da una guaina (mielina) che non solo ha la funzione di protezione, ma contribuisce anche attivamente alla velocità del segnale.
In assenza di mielina il segnale elettrico diventa più lento e debole, rendendo fortemente inefficiente la trasmissione di segnali di attivazione del muscoli così come la ricezione delle informazioni sensoriali dagli arti.
La CMT è causata da mutazioni nel codice genetico, in particolare dove son contenute le istruzioni relative alla produzione di proteine coinvolte nella struttura e nella funzione dell’assone del nervo periferico o della guaina mielinica. Sono più di 40 le porzioni di DNA (geni) potenzialmente coinvolte nelle diverse forme di malattia, anche se più della metà di tutti i casi derivano da una duplicazione del gene PMP22 sul cromosoma 17 (forma CMT1, vide infra).
Ereditarietà e trasmissione
Le mutazioni geniche responsabili della malattia di Charcot-Marie-Tooth sono trasmesse secondo tre diversi modelli:
- Autosomica dominante,
- Autosomica recessiva
- X-Linked dominante (anche detta “legata al cromosoma X”)
- dominante
- recessiva.
Ogni essere umano possiede 23 coppie di cromosomi:
- 22 coppie sono autosomi e vengono ereditate indipendentemente dal sesso biologico della persona (una coppia ereditata da ciascun genitore),
- 1 coppia di eterosomi (cromosomi sessuali), che possono essere
- XX nel caso delle donne, uno ereditato da ciascun genitore,
- XY nel caso degli uomini, con il cromosoma Y ereditato dal padre.
Una malattia è considerata autosomica quando il difetto (mutazione genetica) si trova su uno degli autosomi, e quindi indipendente dal sesso del soggetto:
- È considerata recessiva quando è necessario ereditare il difetto da entrambi i genitori per manifestare la malattia; se ad esempio nessun genitore ne fosse affetto, ma entrambi fossero portatori sani, il rischio di concepire un bambino affetto è del 25%;
- mentre è considerata dominante quando è sufficiente ricevere un’unica copia difettosa, ovvero ad esempio un figlio di un genitore affetto (madre o padre) ed uno sano ha una probabilità del 50% di ereditare la malattia,
Altre forme di CMT sono invece X-linked, che significa che l’errore genetico si trova su un cromosoma sessuale X; anche in questo caso sono note forme trasmesse sia secondo il modello dominante che recessivo (in quest’ultimo caso si verifica una netta prevalenza di pazienti maschi, perché per definizione privi di una potenziale copia sana del gene in questione).
Classificazione
Esistono molte forme di malattia di Charcot-Marie-Tooth, che in generale possono condividere alcuni sintomi, ma che d’altra parte variano profondamente per modello di ereditarietà, età di esordio e cellule coinvolte (assone o guaina).
- La CMT1 è causata da anomalie nella guaina mielinica e si trasmette mediante ereditarietà autosomica dominante; rende conto dell’80% dei casi e può essere ulteriormente distinta in:
- CMT1A, indotta da una duplicazione del gene sul cromosoma 17 che porta le istruzioni per la produzione della proteina mielinica periferica-22 (PMP22), un componente critico della guaina mielinica. La sovraespressione di questo gene (ovvero un’eccessiva produzione di proteine) cause anomalie strutturali e di funzione della guaina. CMT1A decorre in genere in modo progressivo, ma lento, con lo sviluppo di debolezza e atrofia dei muscoli della parte inferiore delle gambe, già a partire dall’infanzia; in seguito compaiono debolezza alla mano, perdita sensoriale e problemi ai piedi e alle gambe.
- La CMT1B è causata da mutazioni nel gene che porta le istruzioni per la produzione della proteina mielinica zero (MPZ o P0), un altro componente critico della guaina mielinica. La maggior parte di queste mutazioni sono di tipo puntiforme, causate cioè da un’unico singolo errore sul codice genetico del DNA (come se nelle pagine di un capitolo di un libro fosse sbagliata una sola lettera). Ad oggi sono state descritte più di 120 diverse mutazioni puntiformi nel gene P0. CMT1B produce sintomi simili a quelli trovati in CMT1A.
- Altre forme meno comuni di CMT1 derivano da mutazioni all’interno di altri geni.
- CMT2 deriva da anomalie nell’assone della cellula nervosa periferica, non più della guaina mielinica come nel caso di CMT1. Poco comune, è una malattia autosomica dominante che può essere distinta in una una dozzina di sottotipi (alcuni dei quali con ulteriori varianti). I sintomi sono simili a quelli osservati nella CMT1, ma i pazienti con CMT2 spesso mostrano una ridotta disabilità e perdita sensoriale. L’esordio solitamente si verifica durante l’infanzia o l’adolescenza. Alcuni tipi di CMT2 possono coinvolgere le corde vocali o il nervo frenico, causando problemi di linguaggio o respirazione.
- La CMT3, o malattia di Dejerine-Sottas, è una neuropatia demielinizzante particolarmente grave che inizia nell’infanzia. I neonati presentano grave atrofia muscolare, debolezza, ritardo nello sviluppo delle capacità motorie e problemi sensoriali. I sintomi possono evolvere in disabilità grave, perdita di sensibilità e curvatura della colonna vertebrale. Può essere causata da mutazioni in più geni e, a seconda dei casi, mediante modello dominante o recessivo.
- CMT4 comprende diversi sottotipi di neuropatie demielinizzanti e assonali e motorie ereditate in modo autosomico recessivo. Ogni sottotipo è causato da una mutazione in un gene diverso, diffusi in modo differente in specifiche etnie e con manifestazioni cliniche caratteristiche. L’esordio è con lo sviluppo di sintomi di debolezza alle gambe durante l’infanzia e nell’adolescenza, in alcuni casi fino alla perdita della capacità di deambulare.
- CMTX1 è la seconda forma più comune di CMT, spesso associata anche ad un’altra forma. Il modello di trasmissione è X-linked:
- i maschi che ereditano il gene mutato mostrano sintomi da moderati a gravi della malattia che iniziano nella tarda infanzia o nell’adolescenza;
- le femmine che ereditano un gene mutato spesso sviluppano sintomi più lievi rispetto ai maschi o non li mostrano affatto.
Sintomi
La CMT colpisce sia i nervi sensoriali (responsabili della raccolta delle sensazioni, come caldo/freddo, pressione, dolore, …) che motori (nervi che attivano un impulso per la contrazione di un muscolo):
- I nervi colpiti degenerano lentamente, perdendo via via la capacità di comunicare con la periferia.
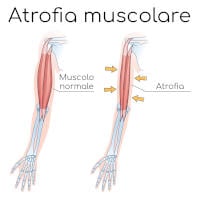
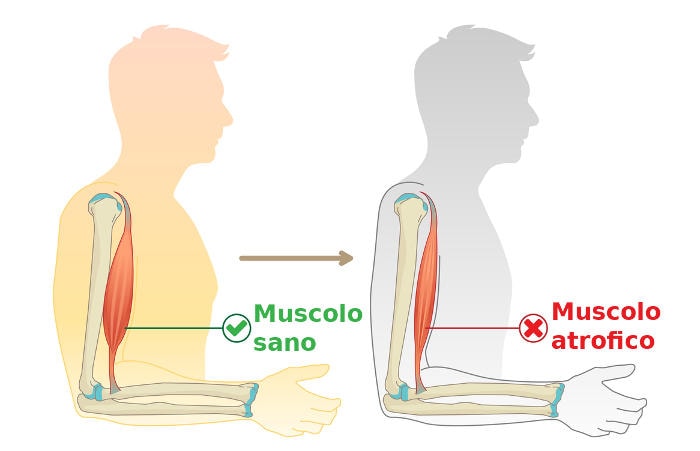
- La degenerazione del nervo motorio provoca debolezza muscolare e diminuzione della massa muscolare (atrofia), che si osserva a livello di braccia, gambe, mani e piedi.
L’esordio in genere riguarda lo sviluppo di debolezza o addirittura paralisi dei muscoli del piede e della parte inferiore della gamba, che si manifestano con la difficoltà a sollevare il piede e con una tipica andatura a gradini, con frequenti inciampi e cadute. Alcuni pazienti lamentano anche problemi di equilibrio.
Anche le deformità del piede, ad esempio un arco pronunciato e dita arricciate (a martello) sono relativamente comuni. La parte inferiore delle gambe può assumere la forma di una “bottiglia di champagne capovolta” a causa della perdita di massa muscolare (ipotrofia ed atrofia).
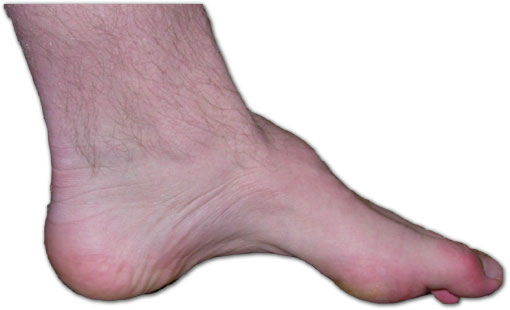
Di Benefros di Wikipedia in inglese – Opera propria (Testo originale: Own work, originally from en.wikipedia; description page is/was here. ), CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=2111831
Con il progredire della malattia possono verificarsi debolezza e atrofia delle mani, che causano non poche difficoltà con le capacità motorie più fini
La degenerazione degli assoni dei nervi sensoriali può invece comportare una ridotta capacità di sentire calore, freddo e più in generale il senso del tatto (la capacità di percezione dei sensi di vibrazione e posizione sono spesso compromessi).
La malattia può anche causare la curvatura della colonna vertebrale (scoliosi) e lo spostamento dell’anca.
Sono comuni le contratture, i crampi muscolari ed un accorciamento cronico dei muscoli e/o dei tendini intorno alle articolazioni, che limitano il movimento.
L’entità del dolore ai nervi (dolore neuropatico) può variare da lieve a grave ed alcuni pazienti potrebbero necessitare di tutori o altri dispositivi ortopedici per mantenere la mobilità.
Sono infine stati descritti
- tremore,
- disturbi della vista e dell’udito.
In rari casi possono verificarsi difficoltà respiratorie in caso di coinvolgimento dei nervi che controllano i muscoli del diaframma.
La progressione dei sintomi è in genere graduale, ma la gravità dei sintomi può variare sensibilmente anche tra gli individui della stessa famiglia.
Diagnosi
La diagnosi della malattia di Charcot-Marie-Tooth inizia con
- anamnesi dettagliata, che indaghi anche la storia familiare,
- un esame neurologico.
Un medico cercherà in particolare evidenze di debolezza muscolare agli arti, diminuzione della massa muscolare, riduzione dei riflessi tendinei e riduzione della sensibilità sensoriale. Possono essere evidenziate deformità del piede e altri problemi ortopedici, come una lieve scoliosi o una formazione anormale dell’articolazione dell’anca.
Un segno piuttosto caratteristico è rappresentato dall’ingrossamento di uno o più nervi, che possono essere avvertiti (o addirittura visti attraverso la pelle), ad esempio a livello del gomito. Questi nervi ingrossati, chiamati nervi ipertrofici, sono il prodotto di guaine mieliniche anormalmente ispessite.
Possono essere richiesti
- studi di conduzione nervosa: alcuni elettrodi vengono posizionati sulla pelle, al di sopra di un muscolo o un nervo. Dietro comando del medico producono un piccolo impulso elettrico che stimola i nervi e fornisce utili informazioni mediante l’analisi dell’attività elettrica da un muscolo o nervo distale;
- Elettromiografia (EMG): prevede l’inserimento di un elettrodo ad ago attraverso la pelle fino al muscolo per la misurazione dell’attività bioelettrica. Anomalie specifiche nelle letture indicano la perdita di assoni. L’EMG può essere utile per caratterizzare ulteriormente la distribuzione, l’attività e la gravità del coinvolgimento dei nervi periferici.
I test genetici, che implicano l’analisi di un campione di sangue, possono rilevare i tipi più comuni di CMT (i test del DNA non sono attualmente disponibili per tutti i tipi di CMT).
Una biopsia del nervo, infine, richiede la rimozione e l’analisi di un piccolo frammento di nervo periferico al microscopio, solitamente prelevato dal polpaccio della gamba attraverso un’incisione nella pelle. Le persone con CMT1 in genere mostrano segni di mielinizzazione anormale.
Cura
In quanto malattia genetica purtroppo ad oggi non esiste una cura per la CMT, ma terapie fisiche e occupazionali, tutori e altri dispositivi ortopedici ed anche la chirurgia ortopedica possono aiutare i pazienti nel far fronte ai sintomi più invalidanti della malattia.
Diversi farmaci antidolorifici possono inoltre essere prescritti in caso di dolore ai nervi.
È molto importante mantenere la mobilità, la flessibilità e la forza muscolare: in questo senso l’inizio precoce di un programma di trattamento può ritardare o ridurre la degenerazione dei nervi e la debolezza muscolare evitando che progredisca fino al punto di disabilità. Possono essere d’aiuto:
- allenamento della forza muscolare,
- stretching (muscolare e dei legamenti),
- esercizio aerobico moderato (attività che aumentino la frequenza cardiaca).
Molte persone con CMT richiedono cavigliere e altri dispositivi ortopedici per mantenere la mobilità quotidiana e prevenire lesioni. I tutori possono aiutare a prevenire le distorsioni della caviglia fornendo supporto e stabilità durante attività come camminare o salire le scale. Anche scarpe o stivali alti possono garantire un utile supporto in caso di debolezza alle caviglie, ma è possibile fare valutazioni simili anche per il pollice della mano.
Alcuni casi selezionati di gravi deformità del piede e delle articolazioni possono beneficiare di un approccio chirurgico, che consente di migliorare la capacità di camminare e ridurre il dolore.
La terapia occupazionale implica l’apprendimento di nuovi approcci e modi per affrontare le attività della vita quotidiana, ad esempio la debolezza delle braccia e delle mani può beneficiare del ricorso a chiusure in velcro e fermagli al posto dei bottoni sui vestiti, così come dispositivi specifici possono consentire un’alimentazione autonoma.
Fonti e bibliografia
Tutti gli aggiornamenti su salute, alimentazione e benessere.