Introduzione
La sindrome di Marfan è una malattia ereditaria del tessuto connettivo che, essendo presente praticamente in qualsiasi distretto anatomico, può manifestarsi in forma di disfunzioni in grado di comparire in qualsiasi organo; i sintomi più comuni sono tuttavia in genere a carico di distretti come
- occhio,
- ossa e articolazioni,
- cuore,
- sistema nervoso centrale,
- vasi sanguigni.
La sindrome è causata da mutazioni per il gene FBN1 presente sul cromosoma 15, che codifica per la proteina fibrillina, espressa in grandi quantità nel tessuto connettivo a cui dona forza e resistenza, offrendo così la giusta integrità strutturale.
I sintomi con cui si manifesta la sindrome possono essere molto variegati e sono rappresentati essenzialmente da:
- articolazioni flessibili,
- dita e mani più lunghe,
- problemi oculari,
- problemi cardiaci (a livello delle valvole e dell’aorta).
La diagnosi è basata sull’anamnesi (presenza di altri casi in famiglia) e sul riconoscimento dei sintomi; è importante un sistematico follow-up con visite specialistiche ed esami strumentali da ripetere annualmente per prevenire l’insorgenza delle complicanze più gravi.
Purtroppo trattandosi di una malattia genetica e quindi presente sin dalla nascita, non esiste alcuna terapia risolutiva, il trattamento mira perciò a prevenire l’insorgenza delle complicanze più gravi, alleviando i sintomi che più gravano sulla qualità di vita del paziente.

iStock.com/Hailshadow
Causa
La sindrome di Marfan è una patologia ereditaria con trasmissione di tipo autosomico dominante: questo significa che una persona affetta ha il 50% di possibilità di trasmettere la malattia ai propri figli.
Nel 20% dei casi tuttavia la mutazione può insorgere spontaneamente in un individuo senza una storia familiare alle spalle.
Ad oggi la sindrome interessa circa 15.000 persone in Italia, con una prevalenza annua di circa 1/5.000 senza differenza tra i sessi.
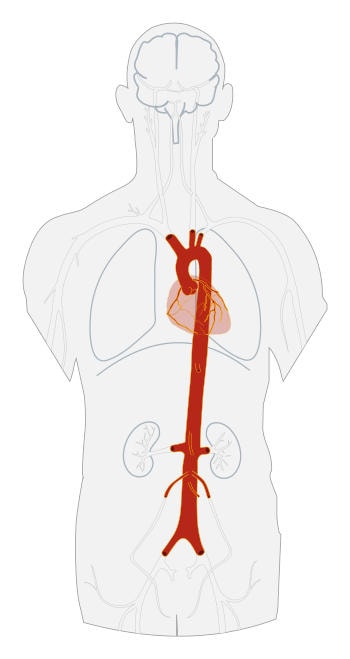
In presenza di un gene della fibrillina mutato, il tessuto connettivo viene alterato, perdendo la sua normale integrità strutturale e a lungo andare provocando la debolezza strutturale di tutti gli organi in cui è presente, ad esempio a livello cardiaco in forma di un indebolimento della parete dell’aorta con sua dilatazione aneurismatica e rottura.
Il tessuto connettivo è fondamentale anche nel collegare ed unire diverse strutture, quindi una sua alterazione può far staccare queste strutture dalle loro inserzioni, come nel caso della retina o del cristallino.
Sintomi
I sintomi possono insorgere a qualsiasi età e variano notevolmente da un paziente all’altro, anche all’interno della stessa famiglia. È possibile osservare un ampio ventaglio di manifestazioni, a partire da sintomi lievi a molto gravi, addirittura con casi di alcuni soggetti che possono rimanere asintomatici sino all’età adulta o per sempre.
Apparato osto-articolare
I soggetti con la Marfan presentano un aspetto longilineo, sono piuttosto magri con un’altezza superiore alla norma rispetto ai loro coetanei; l’apertura delle loro braccia è decisamente aumentata e supera la loro altezza.
Presentano braccia, mani e dita molto lunghe e affusolate, caratteristica quest’ultima, che prende il nome di aracnodattilia, vista la somiglianza delle dita con le zampe dei ragni.
Lo sterno (osso presente al centro del torace, da cui si dipartono le coste), presenta anomalie di struttura con possibilità di avere un torace deformato verso l’interno (petto escavato) o verso l’esterno (petto carenato o torace a botte).
Infine possono aversi piedi piatti, articolazioni ultraflessibili (e spesso dolorose), e deformità della colonna vertebrale che si presenta con cifo-scoliosi.
Cuore e polmoni
I sintomi che si manifestano con problematiche cardio-polmonari sono spesso i più gravi e potenzialmente letali.
L’aorta è il più grasso vaso arterioso dell’organismo, origina dal cuore e si dirama sagittalmente al davanti della colonna vertebrale ramificandosi in altre arterie. Nei soggetti con Marfan la parete del vaso risulta molto indebolita e soggetta quindi a dilatazione sotto la spinta della pressione arteriosa, fino alla formazione di aneurismi; a lungo andare aumenta notevolmente il rischio di rottura dell’aorta (dissecazione aortica) con emorragia gravissima e quasi sempre fatale.
Le valvole cardiache sono in gran parte formate da tessuto connettivo. La loro alterazione strutturale può portare al prolasso valvolare e alla comparsa di insufficienza cardiaca, con rischio di comparsa di aritmie ed endocarditi.
A livello polmonare possono formarsi bolle enfisematose, la cui rottura porta alla comparsa di pneumotorace spontaneo con conseguente dispnea e problemi respiratori.
Occhio
A livello oculare si presenta spesso l’ectopia lentis, ovvero la dislocazione del cristallino che provoca una forte miopia all’individuo.
Ancor più grave può risultare il distacco di retina, che può portare alla cecità permanente se non si ricorre ad intervento chirurgico di urgenza.
Altri sintomi
A livello facciale sono presenti diverse anomalie come la retrognazia mandibolare, il palato ogivale e il massiccio facciale allungato verticalmente: queste alterazioni portano ad una alterazione nello sviluppo del linguaggio e vanno a rappresentare la cosiddetta facies marfanoide.
Diagnosi
Identificare la sindrome di Marfan può essere complesso poiché i segni clinici variano sensibilmente tra gli individui e si sovrappongono ad altre patologie del tessuto connettivo. La diagnosi moderna si basa sull’applicazione rigorosa della Nosologia di Ghent revisionata, uno standard internazionale che combina parametri clinici, storia familiare e test genetici.
Esame clinico e punteggio sistemico
Il medico valuta la presenza di caratteristiche fisiche specifiche attraverso un “punteggio sistemico”. Vengono analizzati segni come la deformità del torace (petto carinato o escavato), il segno del polso e del pollice (che indicano l’aracnodattilia), la scoliosi e la ridotta estensione dei gomiti. Un punteggio uguale o superiore a 7, associato ad altri criteri chiave, orienta con decisione verso la diagnosi.
Valutazione cardiologica e oculistica
Due pilastri fondamentali della diagnosi sono:
- Ecocardiografia: l’ecografia del cuore è l’esame di elezione per misurare il diametro della radice aortica. Si utilizza il “Z-score”, un parametro che confronta le dimensioni dell’aorta del paziente con quelle attese per età e superficie corporea. La rilevazione di una dilatazione significativa o di un prolasso valvolare è un criterio diagnostico maggiore.
- Esame alla lampada a fessura: una visita oculistica approfondita è necessaria per rilevare l’ectopia lentis (dislocazione del cristallino), segno distintivo della sindrome presente in circa l’80% dei casi.
Test genetici
L’analisi del DNA per identificare mutazioni nel gene FBN1 è oggi uno strumento standard. Sebbene non tutti i pazienti con mutazione FBN1 abbiano la sindrome di Marfan, e non tutti i pazienti con diagnosi clinica presentino una mutazione identificabile (per limiti tecnici del test), la genetica è cruciale per confermare casi dubbi e per lo screening dei familiari. La consulenza genetica è raccomandata per interpretare correttamente i risultati e discutere i rischi di trasmissione.
Diagnosi differenziale e imaging avanzato
È essenziale escludere patologie simili come la sindrome di Loeys-Dietz o la sindrome di Ehlers-Danlos. In alcuni casi, il medico può richiedere una Angio-TC o una Risonanza Magnetica (RM) per visualizzare l’intera aorta e non solo la parte vicina al cuore, garantendo una mappatura completa di eventuali aneurismi.
Cura
Sebbene non esista una cura definitiva per correggere l’anomalia genetica, la gestione terapeutica moderna ha trasformato radicalmente la prognosi della sindrome di Marfan, permettendo un’aspettativa di vita sovrapponibile a quella della popolazione generale. L’obiettivo primario è la prevenzione delle complicanze aortiche e la protezione della vista e della funzionalità scheletrica.
Terapia farmacologica
La protezione del sistema cardiovascolare inizia con la gestione della pressione arteriosa per ridurre lo stress sulle pareti dell’aorta.
I protocolli attuali prevedono l’uso di:
- Beta-bloccanti: rappresentano la prima scelta per rallentare la frequenza cardiaca e la forza di contrazione, riducendo la velocità di dilatazione dell’aorta.
- Sartani (Antagonisti del recettore dell’angiotensina II): farmaci come il losartan hanno dimostrato un’efficacia paragonabile o complementare ai beta-bloccanti, agendo anche su segnali chimici cellulari che contribuiscono alla degradazione del tessuto connettivo. Spesso vengono utilizzati in combinazione per massimizzare la protezione.
Gestione chirurgica
L’intervento chirurgico preventivo sull’aorta è un pilastro del trattamento. Viene raccomandato quando il diametro della radice aortica raggiunge una soglia critica (generalmente tra 45 e 50 mm, a seconda dei fattori di rischio individuali). Le tecniche includono la sostituzione del tratto dilatato con una protesi sintetica, preferendo quando possibile procedure “valve-sparing” che conservano la valvola aortica originale del paziente, riducendo la necessità di terapie anticoagulanti croniche.
Trattamenti per ossa e occhi
La gestione ortopedica può includere l’uso di corsetti per la scoliosi durante la crescita o la correzione chirurgica in casi gravi. Per quanto riguarda la vista, la miopia elevata viene corretta con lenti o contatti, mentre l’ectopia lentis o il distacco di retina richiedono interventi microchirurgici specialistici per preservare la capacità visiva.
Stile di vita e prevenzione
Le modifiche dello stile di vita sono determinanti per la sicurezza del paziente. Si raccomanda di evitare sport di contatto (calcio, arti marziali) e attività isometriche intense (sollevamento pesi), che causano bruschi picchi di pressione arteriosa. Sono invece incoraggiate attività aerobiche leggere come il nuoto o la camminata veloce.
Un’attenzione particolare è rivolta alla gravidanza, che rappresenta un periodo ad alto rischio per la dissecazione aortica; le donne affette devono essere seguite in centri specializzati per gravidanze ad alto rischio, con un monitoraggio ecocardiografico frequente.
L’approccio multidisciplinare
Data la complessità della sindrome, il paziente viene preso in carico da un team coordinato che comprende:
- Cardiologo e cardiochirurgo.
- Genetista clinico.
- Oftalmologo.
- Ortopedico e fisiatra.
- Psicologo, fondamentale per gestire l’impatto di una malattia cronica ereditaria.
La prognosi è oggi estremamente favorevole se la diagnosi è precoce e il monitoraggio è costante. La chiave del successo terapeutico risiede nell’aderenza ai controlli periodici (ecocardiogramma annuale o semestrale) e alla terapia farmacologica prescritta, fattori che riducono drasticamente il rischio di eventi acuti.
Fonti e bibliografia
Tutti gli aggiornamenti su salute, alimentazione e benessere.